Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Vicky Zhou and Version 3 by Jacopo Umberto Verga.
Exposure to Endocrine Disrupting Chemicals (EDCs) has been linked with several adverse outcomes. Transcriptome-wide analyses using RNAseq provide snapshots of cellular, tissue, and whole organism transcriptomes under normal physiological and EDC perturbed conditions. A global view of gene expression provides highly valuable information as it uncovers gene families or more specifically, pathways that are affected by EDC exposures, but also reveals those that are unaffected. Hypotheses about genes with unknown functions can also be formed by comparison of their expression levels with genes of known function.
- Endocrine Disrupting Chemicals (EDC)
- DNA microarrays
- RNA sequencing
- Adverse Outcome Pathway (AOP)
- Molecular Initiating Event (MIE)
- Key Events (KE)
- risk assessment
1. Introduction
In the twentieth century a large quantity of contaminants, both organic and inorganic, has been released into the environment . Many of these industrial chemicals have sufficient structural similarity to steroid hormones to be able to bind to steroid receptors or enzymes that regulate steroid hormone concentrations and, thus, perturb normal endocrine physiology in aquatic species, animals, and humans [1][2][3][4][5][6]. Those chemicals capable of interfering with the endocrine system, mimicking the action of endogenous messengers (hormones) with their specific receptors are defined as endocrine-disrupting chemicals (ECDs) [7]. This exposure of humans, fish, and other animals to Endocrine DCs isrupting Chemicals (EDCs) is a global concern and the subject of regulatory activity by various environmental agencies [3][8].
EDCs interfere with many hormone-regulated physiological pathways and have complex effects on human and fish physiology due to the diversity of the hormone receptors and enzymes that they bind [9]. The detailed mechanisms by which ED compounds act is not completely understood, but many interact with nuclear receptors, such as the retinoid X receptor (RXR) [10][11][12][13], peroxisome proliferator-activated receptors (PPARs) [13][14][15][16][17] and estrogen receptors (ERs) [18][19]. The binding of EDCs to these and other receptors, as well as steroidogenic enzymes, can perturb normal physiological processes. The wide range of health effects attributed to EDCs include reproductive dysfunction (e.g., distorted male/female sex ratios in fish, elevated or reduced hormone levels, diminished fertility, and male and female reproductive tract abnormalities) [20][21]; premature puberty [22]; neurological [23][24] and behavioral [25][26][27][28] complications; immune dysregulation [29][30]; cancer [31][32][33][34][35] and metabolic diseases [29][36][37][38][39][40][41][42][43][44][45]. EDCs include personal care products, plasticizers, detergents, insecticides, and pharmaceuticals. Many of these chemicals enter rivers and the ocean, where they directly interact with aquatic life and disrupt endocrine responses. Humans are exposed to these EDCs from drinking water and dietary consumption of fish [46].
2. Genomics and Bioinformatics Approaches in Endocrine Disruptor Research
Over the past two decades, the field of ecotoxicology has adapted genomics technologies to better understand how exposure to compounds perturbs gene expression on a molecular level, facilitating transcriptome analysis. Historically, two tools have been used for this purpose: DNA microarrays and RNA sequencing (RNA-Seq) although microarrays are becoming an obsolete technology [47]. A challenge is to comprehend the effects of these chemicals on the health of humans as well as other animals. Much research has been carried out elucidating what concentrations of EDCs are toxic for regulatory purposes [48][49]. Critical for obtaining this information is the identification of biomarkers and genomic signatures of the response to different chemicals in humans, fish, and other animals. This is a challenging task because the actions of each chemical are complex, involving the regulation of many genes and interactions between many physiological pathways. The methods for investigating the molecular effects of exposure to EDCs must sample a broad molecular response. A special concern regarding the effects of EDCs is that exposure at different stages in life: embryonic, postnatal, juvenile, and adult are likely to have different effects. Thus, although molecular analyses taken soon after exposure to a chemical are useful in diagnosis the response to a chemical, it also is necessary to perform these analyses over a generation and if possible, for more than one generation, to uncover delayed effects. A good example is the diethylstilbestrol (DES) exposure which manifests in cancer development many years following the exposure and even affects later generations[50].
Genomics technologies have enabled the assessment of the effects of xenobiotics in the environment on human health [51]. Ecological exposure and risk assessment models alone, however, are not adequate to examine the effect of EDCs [51]. Producing exposure-to-outcome databases from diverse ecotoxicogenomic datasets and executing systems toxicology approaches, are required to comprehensively explain the risk of these toxicants and to assuage the ambiguities that currently exist with risk assessment. Recent work in sediment and dredged material assessment has advocated for the inclusion of biomarkers in acute bioassays [52]. Although chemical analyses have enabled the development of standards for regulating contaminant occurrence in the environment, there is a need for sensitive assays that can detect their bioavailability in fish, animals, and humans. Chronic exposure to compounds with endocrine targets, particularly at low chemical levels, has been shown to disrupt vertebrate development [2][5][53][54]. Recent advances in molecular methods, have substantially improved the sensitivity for assessing the biological effects of chemical contaminants of emerging concern (CECs) on animal physiology [1][55][56][57], but more pertinent, these tools provide chemical signatures of bioavailability and exposure and allow the development of risk assessment databases.
2.1. RNA Sequencing
From 2005, several massively parallel sequencing technologies termed Next-Generation Sequencing (NGS) emerged, resulting in increased throughput and accuracy, and reducing sequencing costs to less than a thousand dollars for a human genome. These high throughput parallel DNA sequencing platforms launched a new era of genomics and molecular biology. A key application is RNA sequencing (RNA-Seq) [58]. With RNA-Seq, the RNA is extracted from the biological samples and converted to cDNA [58] capturing the sequences and abundance of all mRNA transcripts at a given time point. RNA-Seq can be used to obtain per-base expression profiles [58], with several advantages when compared with microarrays including better sensitivity for those genes that are expressed at low or very high levels, splice variants, and non-coding transcripts (such as miRNA and lncRNA) [58][59].
Microarray analyses of the transcriptome have waned in the past five years as there are limitations to this method compared with RNAseq: the inability to detect novel transcripts that do not have specifically designed probes, the appearance of cross-hybridization artifacts in analyses of similar sequences, and decreased accuracy for transcripts present only at low levels [60][61][62][63][64][65][66]. Sequencing-based approaches to transcriptome profiling overcome these issues and are designed to directly determine transcript sequences [62]. In comparison to microarrays, RNA-Seq has minimal background technical noise. The sequence reads can be unequivocally mapped to unique regions in the genome and the technology has a greater dynamic range to quantify gene expression level. RNA-Seq can produce highly reproducible results, leveraging both biological and technological replicates, and requires less input RNA material [67][68][69].
Some of the drawbacks of RNAseq have been centered on data management, ease of use, and the total number of databases provided by both the scientific community and commercial vendors [70]. HTS experiments generate FASTQ files, massive raw data files containing the nucleotide sequence, and quality score information. Considered “raw” data, FASTQ files are subject to secondary analysis, often including alignment to a reference genome or de-novo assembly, which generates secondary and intermediate files as massive as the initial, “raw” data. These derived files, in turn, may be stored, filtered, annotated, or analyzed in several ways that generate more data; in short, storage and organization of RNA-Seq data provide a challenge. In terms of the user experience, many current HTS tools require knowledge of intricate command-line instructions for their operation. This provides a considerable barrier for nontechnical audiences—who have little experience in computer science—from effectively utilizing RNA-Seq for their analyses. Finally, the number of databases and knowledge bases is notoriously expansive and increases in size every year; the most recent count, according to Nucleic Acids Research, is 1641 databases as of January 2021 [71]. Keeping up with these databases is a daunting challenge, and recent efforts have focused on improving these standards of knowledge sharing [72].
2.2. RNAseq for the Study of Endocrine Disruption
Leet et al. investigated the effects of early life exposures to the herbicide atrazine or EE2 on sexual differentiation and gene expression in gonadal tissue [73]. They used largemouth bass (Micropterus salmoides) from 7 to 80 days post-spawn to concentrations of 1, 10, or 100 µg atrazine/L or 1 or 10 ng EE2/L and monitored histological development and transcriptomic changes in gonad tissue. They noted an almost 100% female sex ratio in fish exposed to EE2 at 10 ng/L, likely as a result of sex reversal of males [73]. Not surprisingly many gonad genes were differentially expressed between the sexes.
Wang et al. examined the effects of 17α-methyltestosterone (MT), an artificial androgenic compound, used to induce masculinization of both secondary sex characteristics and gonads in aquatic studies. The Stone moroko (Pseudorasbora parva) was exposed to MT, and the growth and development of fish were delayed by exposure to MT at 200 ng/L. RNAseq analyses revealed 7758 and 11,543 DEGs in females and males. MT had more obvious disruption effects on males than females, and this was primarily reflected in the immune system [74].
Renaud et al., examined the effects of EE2 exposure on the Pacific sardine (Sardinops sagax) and chub mackerel (Scomber japonicus). RNA sequencing (RNAseq) was performed on liver RNA harvested from wild sardine and mackerel exposed for 5 h under laboratory conditions to a concentration of 12.5 pM EE2. This revealed that environmental levels of EE2 disrupted basic biological processes and pathways in both male sardine and mackerel, leading to molecular signatures of metabolic, hormonal, and immune dysfunction, as well as carcinogenesis in exposed fish [75].
Bertucci and colleagues examined chronic exposure to wastewater treatment plant and stormwater effluents at the whole-transcriptome level in the Asian clam (Corbicula fluminea) and evaluated the physiological outcomes. They uncovered a set of 3181 transcripts with altered abundance in response to water quality. The largest differences in transcriptomic profiles were observed between clams from the reference clean site and those exposed to wastewater treatment plant effluents. Most of the differentially expressed transcripts were involved in signaling pathways involved in energy metabolism suggesting an energy/nutrient deficit and hypoxic conditions in response to the pollutants in the effluents [76].
Legrand and co-workers exposed copepods (Eurytemora affinis) to sublethal concentrations of the pesticide pyriproxyfen (PXF) and insecticide chlordecone (CLD). After 48 h, males and females (400 individuals each) were sorted for RNA extraction. In total, 2566 different genes were differentially expressed after EDC exposures compared to controls with similar numbers of DE genes with both compounds. More genes were differentially expressed in males than in females after both exposures [77].
Guo et al. evaluated the effects of effluents containing phenolic compounds from the Ba River on the ovary of the Sharpbelly (Hemiculter leucisculus), a freshwater fish using transcriptomic and metabolomic analyses. In fish collected near wastewater discharge, oocyte development was activated, compared to upstream and remote sites. Histopathological alterations were found in the fish ovaries likely a result of upregulated steroid hormone biosynthesis, as suggested by the differentially expressed genes from the RNAseq [78].
3. The Adverse Outcomes Framework
The Adverse Outcomes Framework (AOF), developed in 2007 with the release of a National Research Council report entitled “Toxicity Testing in the 21st Century,” has become a popular model in the study of toxicology. This model defines an Adverse Outcome Pathway (AOP) as a linear pathway comprised of a molecular initiation event (MIE), key events (KE), and the Adverse Outcome (AO) itself causally linked together (Figure 1). Using cancer as an example, the MIE would be a single mutation in a gene associated with, for example, control of the cell cycle. This, alone, does not cause cancer but may trigger pathway-level perturbations, so-called key events, that could reduce the expression of tumor-suppression genes and mutate proto-oncogenes into oncogenes. The goal of this framework is to transform current toxicology testing by supporting less animal-intensive alternatives to toxicity testing and predictive ecotoxicology [79][80].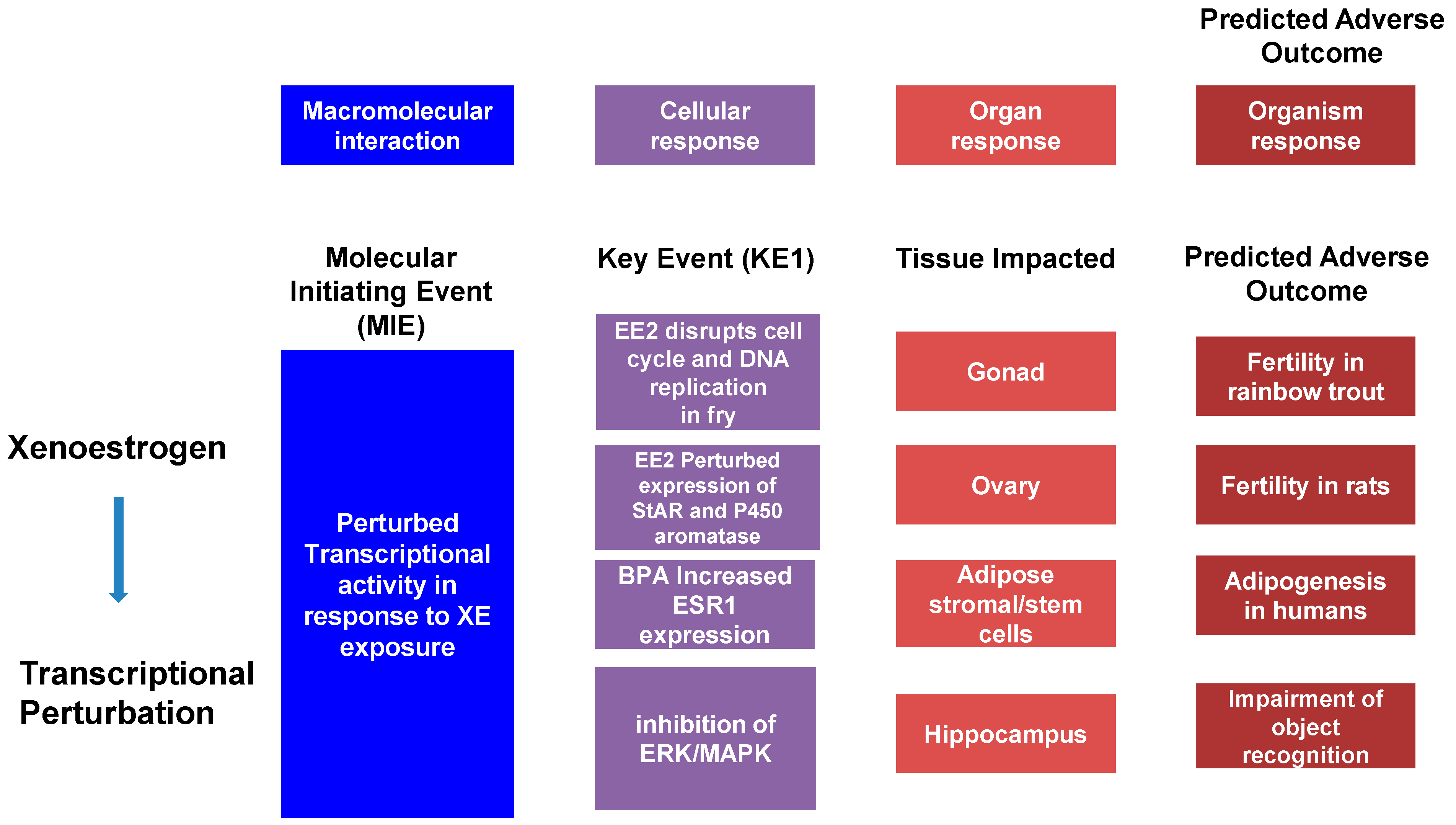
Figure 1. The adverse outcome pathway (AOP) is a linear pathway composed of a Molecular Initiating Event (MIE), Key Events (KE), and an Adverse Outcome (AO) causally linked together. Example AOPs are illustrated.
Adverse Outcomes are not always the result of a single event. Cancer, for instance, is more common among adults as they age due to genomic damage including point mutations and insertion-deletions that disrupt normal cell function, allowing for the uncontrolled growth and division of cells. The first of these mutations is defined as the MIE, which is then followed by other mutations—KE—that are linked by KE relationships (KERs) and further alter expression in such a way that an adverse outcome is the result. Going beyond the simple, linear AOPs are AOP Networks, quantitative AOPs (qAOPs), and qAOP Networks. An AOP network is a system of interacting AOPs with shared KEs, while qAOPs are AOPs in which the KERs (qKERs) are given a weight/value that quantitatively characterizes the relationship between KEs in an AOP. The AOF identifies qAOPs as the “ideal” form to be used in risk assessment due to this quantification [79][80].
To help make the AOF easier to comprehend, Villeneuve et al., developed a set of principles that summarize the development of this model [81]: (1) because any external stress or chemical that can trigger an MIE has the potential to activate a chain of KEs leading to the AOP; AOPs are not specific to given chemicals. (2) AOPs are composed of two fundamental units: KEs and KERs that are usually shared between AOPs. (3) AOPs are not a thorough interpretation of biological processes, but a basic and organized framework for organizing toxicological evidence; this defines individual AOPs as rational units of AOP development. (4) Those networks compiled of numerous AOPs are likely the most functional units of prediction in everyday scenarios, as the prediction of adverse outcomes will often involve contemplation of multiple AOPs. Finally, (5) AOPs are not stationary and will evolve as new insights are offered [79][81].
As the AOP is a developing concept there are some limitations to the framework that must be addressed to improve its viability. One of the most critical considerations is the ability of AOPs to accurately define scientifically robust connections between the MIE, KEs, and adverse outcomes [79]. The application of AOP networks and qAOPs, described above, will facilitate more accurate predictions, but at present, there remain gaps between these events. It has been suggested that, for AOPs to be useful as more than a categorizing tool, they must increase trust for risk decisions more than existing approaches, and must be able to reduce animal usage as well [80]. In short, the AOP framework will need to demonstrate, beyond a shadow of a doubt, that it is effective at predicting adverse outcomes in order for it to be accepted by the scientific community [79].
As outlined above, EDCs have become common in the environment; though there are efforts to reduce these compounds humans are still potentially exposed to these chemicals. The studies above utilize two approaches for determining the toxicity of these EDCs: using toxicological concentrations of XEs or using levels found in the environment, in other words, the concentrations most people will be exposed to daily. The AOP framework establishes that the development of an adverse outcome is not necessarily completed overnight; rather, a linear series of key events following the initial MIE create a stockpile of genetic damage that, later in life, impact expression in such a way that the adverse outcome is the result.
4. Conclusions
To conclude, endocrine disrupting compounds are ubiquitous in modern life. BPA, for example, is an industrial chemical used in the fabrication of plastics and found in the urine samples of up to 96% of Americans [82]. It is estimated that 1000 of the 85,000 synthetic chemicals in existence may be EDCs, however, the majority of these have not undergone sufficient testing to confirm this, so the true number may be much higher [83]. In addition, there are a large number of chemicals that have yet to be fully profiled to determine their capacity to act as EDCs. The cost of profiling each chemical is not insignificant. Genomic and bioinformatics approaches have revolutionized toxicology testing, permitting deep insights into the transcriptomes of cells and tissues following exposure to EDCs and reducing the costs that are associated with traditional toxicity testing. Genomic profiling linked to computational approaches, such as The Open TG-GATES database presents an opportunity to avoid the cost associated with profiling each chemical for its efficacy as an EDC and to accelerate the process using high-throughput informatics [84][85]. The Adverse Outcome Pathway (AOP) framework integrates these genomic and computational outputs and allows a molecular initiation event, key events resulting from this initial insult, and the Adverse Outcome (AO) itself to be causally linked together in the context of predictive toxicology assessments.
References
- Chen, G.; White, P.A. The Mutagenic Hazards of Aquatic Sediments: A Review. Mutat. Res. 2004, 567, 151–225.
- Baker, M.E.; Ruggeri, B.; Sprague, L.J.; Eckhardt-Ludka, C.; Lapira, J.; Wick, I.; Soverchia, L.; Ubaldi, M.; Polzonetti-Magni, A.M.; Vidal-Dorsch, D.; et al. Analysis of Endocrine Disruption in Southern California Coastal Fish Using an Aquatic Multispecies Microarray. Environ. Health Perspect. 2009, 117, 223–230.
- Grün, F.; Watanabe, H.; Zamanian, Z.; Maeda, L.; Arima, K.; Cubacha, R.; Gardiner, D.M.; Kanno, J.; Iguchi, T.; Blumberg, B. Endocrine-Disrupting Organotin Compounds Are Potent Inducers of Adipogenesis in Vertebrates. Mol. Endocrinol. 2006, 20, 2141–2155.
- Lam, S.H.; Mathavan, S.; Tong, Y.; Li, H.; Karuturi, R.K.M.; Wu, Y.; Vega, V.B.; Liu, E.T.; Gong, Z. Zebrafish Whole-Adult-Organism Chemogenomics for Large-Scale Predictive and Discovery Chemical Biology. PLoS Genet. 2008, 4, e1000121.
- Lange, A.; Paull, G.C.; Coe, T.S.; Katsu, Y.; Urushitani, H.; Iguchi, T.; Tyler, C.R. Sexual Reprogramming and Estrogenic Sensitization in Wild Fish Exposed to Ethinylestradiol. Environ. Sci. Technol. 2009, 43, 1219–1225.
- Soto, A.M.; Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C. Does Breast Cancer Start in the Womb? Basic Clin. Pharmacol. Toxicol. 2008, 102, 125–133.
- Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C.; Rubin, B.S.; Soto, A.M. Bisphenol-A and the Great Divide: A Review of Controversies in the Field of Endocrine Disruption. Endocr. Rev. 2009, 30, 75–95.
- La Merrill, M.A.; Vandenberg, L.N.; Smith, M.T.; Goodson, W.; Browne, P.; Patisaul, H.B.; Guyton, K.Z.; Kortenkamp, A.; Cogliano, V.J.; Woodruff, T.J.; et al. Consensus on the Key Characteristics of Endocrine-Disrupting Chemicals as a Basis for Hazard Identification. Nat. Rev. Endocrinol. 2020, 16, 45–57.
- Vajda, A.M.; Barber, L.B.; Gray, J.L.; Lopez, E.M.; Woodling, J.D.; Norris, D.O. Reproductive Disruption in Fish Downstream from an Estrogenic Wastewater Effluent. Environ. Sci. Technol. 2008, 42, 3407–3414.
- Toporova, L.; Balaguer, P. Nuclear Receptors Are the Major Targets of Endocrine Disrupting Chemicals. Mol. Cell. Endocrinol. 2020, 502, 110665.
- Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. RXRalpha, PXR and CAR Xenobiotic Receptors Mediate the Apoptotic and Neurotoxic Actions of Nonylphenol in Mouse Hippocampal Cells. J. Steroid Biochem. Mol. Biol. 2016, 156, 43–52.
- Huang, W.; Wu, Q.; Xu, F.; Li, L.; Li, J.; Que, H.; Zhang, G. Functional Characterization of Retinoid X Receptor with an Emphasis on the Mediation of Organotin Poisoning in the Pacific Oyster (Crassostrea gigas). Gene 2020, 753, 144780.
- Inderbinen, S.G.; Engeli, R.T.; Rohrer, S.R.; Di Renzo, E.; Aengenheister, L.; Buerki-Thurnherr, T.; Odermatt, A. Tributyltin and Triphenyltin Induce 11β-Hydroxysteroid Dehydrogenase 2 Expression and Activity through Activation of Retinoid X Receptor α. Toxicol. Lett. 2020, 322, 39–49.
- Milton, F.A.; Lacerda, M.G.; Sinoti, S.B.P.; Mesquita, P.G.; Prakasan, D.; Coelho, M.S.; de Lima, C.L.; Martini, A.G.; Pazzine, G.T.; Borin, M.d.F.; et al. Dibutyltin Compounds Effects on PPARγ/RXRα Activity, Adipogenesis, and Inflammation in Mammalians Cells. Front. Pharm. 2017, 8, 507.
- Lopes, C.; Madureira, T.V.; Ferreira, N.; Pinheiro, I.; Castro, L.F.; Rocha, E. Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) in Brown Trout: Interference of Estrogenic and Androgenic Inputs in Primary Hepatocytes. Environ. Toxicol. Pharm. 2016, 46, 328–336.
- Fang, H.; Fang, W.; Cao, H.; Luo, S.; Cong, J.; Liu, S.; Pan, F.; Jia, X. Di-(2-Ethylhexyl)-Phthalate Induces Apoptosis via the PPARγ/PTEN/AKT Pathway in Differentiated Human Embryonic Stem Cells. Food Chem. Toxicol. 2019, 131, 110552.
- Shoaito, H.; Petit, J.; Chissey, A.; Auzeil, N.; Guibourdenche, J.; Gil, S.; Laprévote, O.; Fournier, T.; Degrelle, S.A. The Role of Peroxisome Proliferator–Activated Receptor Gamma (PPARγ) in Mono(2-Ethylhexyl) Phthalate (MEHP)-Mediated Cytotrophoblast Differentiation. Environ. Health Perspect. 2019, 127, 27003.
- Parillo, F.; Maranesi, M.; Brecchia, G.; Gobbetti, A.; Boiti, C.; Zerani, M. In Vivo Chronic and in Vitro Acute Effects of Di(2-Ethylhexyl) Phthalate on Pseudopregnant Rabbit Corpora Lutea: Possible Involvement of Peroxisome Proliferator-Activated Receptor Gamma. Biol. Reprod. 2014, 90, 41.
- Kim, Y.S.; Hwang, K.A.; Hyun, S.H.; Nam, K.H.; Lee, C.K.; Choi, K.C. Bisphenol A and Nonylphenol Have the Potential to Stimulate the Migration of Ovarian Cancer Cells by Inducing Epithelial-Mesenchymal Transition via an Estrogen Receptor Dependent Pathway. Chem. Res. Toxicol. 2015, 28, 662–671.
- Kitraki, E.; Nalvarte, I.; Alavian-Ghavanini, A.; Ruegg, J. Developmental Exposure to Bisphenol A Alters Expression and DNA Methylation of Fkbp5, an Important Regulator of the Stress Response. Mol. Cell. Endocrinol. 2015, 417, 191–199.
- Jiang, J.; Ma, L.; Yuan, L.; Wang, X.; Zhang, W. Study on Developmental Abnormalities in Hypospadiac Male Rats Induced by Maternal Exposure to Di-n-Butyl Phthalate (DBP). Toxicology 2007, 232, 286–293.
- Liu, K.; Lehmann, K.P.; Sar, M.; Young, S.S.; Gaido, K.W. Gene Expression Profiling Following in Utero Exposure to Phthalate Esters Reveals New Gene Targets in the Etiology of Testicular Dysgenesis. Biol. Reprod. 2005, 73, 180–192.
- Fudvoye, J.; Lopez-Rodriguez, D.; Franssen, D.; Parent, A.-S. Endocrine Disrupters and Possible Contribution to Pubertal Changes. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101300.
- Saaristo, M.; Wong, B.B.; Mincarelli, L.; Craig, A.; Johnstone, C.P.; Allinson, M.; Lindstrom, K.; Craft, J.A. Characterisation of the Transcriptome of Male and Female Wild-Type Guppy Brains with RNA-Seq and Consequences of Exposure to the Pharmaceutical Pollutant, 17alpha-Ethinyl Estradiol. Aquat. Toxicol. 2017, 186, 28–39.
- Komada, M.; Gendai, Y.; Kagawa, N.; Nagao, T. Prenatal Exposure to Di(2-Ethylhexyl) Phthalate Impairs Development of the Mouse Neocortex. Toxicol. Lett. 2016, 259, 69–79.
- Braun, J.M.; Kalkbrenner, A.E.; Calafat, A.M.; Yolton, K.; Ye, X.; Dietrich, K.N.; Lanphear, B.P. Impact of Early-Life Bisphenol A Exposure on Behavior and Executive Function in Children. Pediatrics 2011, 128, 873–882.
- Wang, C.; Li, Z.; Han, H.; Luo, G.; Zhou, B.; Wang, S.; Wang, J. Impairment of Object Recognition Memory by Maternal Bisphenol A Exposure Is Associated with Inhibition of Akt and ERK/CREB/BDNF Pathway in the Male Offspring Hippocampus. Toxicology 2016, 341–343, 56–64.
- Chen, F.; Zhou, L.; Bai, Y.; Zhou, R.; Chen, L. Hypothalamic-Pituitary-Adrenal Axis Hyperactivity Accounts for Anxiety- and Depression-like Behaviors in Rats Perinatally Exposed to Bisphenol A. J. Biomed. Res. 2015, 29, 250–258.
- Yu, C.J.; Du, J.C.; Chiou, H.C.; Yang, S.H.; Liao, K.W.; Yang, W.; Chung, M.Y.; Chien, L.C.; Hwang, B.; Chen, M.L. Attention Deficit/Hyperactivity Disorder and Urinary Nonylphenol Levels: A Case-Control Study in Taiwanese Children. PLoS ONE 2016, 11, e0149558.
- Morii, K.; Nishisaka, M.; Nakamura, S.; Oda, T.; Aoyama, Y.; Yamamoto, T.; Kishida, H.; Okushin, H.; Uesaka, K. A Case of Synthetic Oestrogen-Induced Autoimmune Hepatitis with Microvesicular Steatosis. J. Clin. Pharm. 2014, 39, 573–576.
- Tao, J.; Sun, L.X.; Le, X.C. Study of the Effects of Bisphenol A Using Human Fetal Lung Fibroblasts. J. Environ. Sci. 2016, 48, 6–10.
- Koike, E.; Yasuda, Y.; Shiota, M.; Shimaoka, M.; Tsuritani, M.; Konishi, H.; Yamasaki, H.; Okumoto, K.; Hoshiai, H. Exposure to Ethinyl Estradiol Prenatally and/or after Sexual Maturity Induces Endometriotic and Precancerous Lesions in Uteri and Ovaries of Mice. Congenit. Anom. 2013, 53, 9–17.
- Hu, W.Y.; Shi, G.B.; Hu, D.P.; Nelles, J.L.; Prins, G.S. Actions of Estrogens and Endocrine Disrupting Chemicals on Human Prostate Stem/Progenitor Cells and Prostate Cancer Risk. Mol. Cell. Endocrinol. 2012, 354, 63–73.
- Oral, D.; Erkekoglu, P.; Kocer-Gumusel, B.; Chao, M.W. Epithelial-Mesenchymal Transition: A Special Focus on Phthalates and Bisphenol A. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 43–58.
- Zhu, M.; Huang, C.; Ma, X.; Wu, R.; Zhu, W.; Li, X.; Liang, Z.; Deng, F.; Wu, J.; Geng, S.; et al. Phthalates Promote Prostate Cancer Cell Proliferation through Activation of ERK5 and P38. Environ. Toxicol. Pharm. 2018, 63, 29–33.
- Park, M.-A.; Hwang, K.-A.; Lee, H.-R.; Yi, B.-R.; Jeung, E.-B.; Choi, K.-C. Cell Growth of BG-1 Ovarian Cancer Cells Is Promoted by Di-n-Butyl Phthalate and Hexabromocyclododecane via Upregulation of the Cyclin D and Cyclin-Dependent Kinase-4 Genes. Mol. Med. Rep. 2012, 5, 761–766.
- Grun, F.; Blumberg, B. Perturbed Nuclear Receptor Signaling by Environmental Obesogens as Emerging Factors in the Obesity Crisis. Rev. Endocr. Metab. Disord. 2007, 8, 161–171.
- Marmugi, A.; Lasserre, F.; Beuzelin, D.; Ducheix, S.; Huc, L.; Polizzi, A.; Chetivaux, M.; Pineau, T.; Martin, P.; Guillou, H.; et al. Adverse Effects of Long-Term Exposure to Bisphenol A during Adulthood Leading to Hyperglycaemia and Hypercholesterolemia in Mice. Toxicology 2014, 325, 133–143.
- Li, J.; Lai, H.; Chen, S.; Zhu, H.; Lai, S. Gender Differences in the Associations between Urinary Bisphenol A and Body Composition among American Children: The National Health and Nutrition Examination Survey, 2003–2006. J. Epidemiol. 2017, 27, 228–234.
- Luzio, A.; Monteiro, S.M.; Rocha, E.; Fontainhas-Fernandes, A.A.; Coimbra, A.M. Development and Recovery of Histopathological Alterations in the Gonads of Zebrafish (Danio rerio) after Single and Combined Exposure to Endocrine Disruptors (17alpha-Ethinylestradiol and Fadrozole). Aquat. Toxicol. 2016, 175, 90–105.
- Young, B.J.; Lopez, G.C.; Cristos, D.S.; Crespo, D.C.; Somoza, G.M.; Carriquiriborde, P. Intersex and Liver Alterations Induced by Long-Term Sublethal Exposure to 17alpha-Ethinylestradiol in Adult Male Cnesterodon decemmaculatus (Pisces: Poeciliidae). Environ. Toxicol. Chem. 2016, 36, 1738–1745.
- Bernabo, I.; Biasone, P.; Macirella, R.; Tripepi, S.; Brunelli, E. Liver Histology and Ultrastructure of the Italian Newt (Lissotriton Italicus): Normal Structure and Modifications after Acute Exposure to Nonylphenol Ethoxylates. Exp. Toxicol. Pathol. 2014, 66, 455–468.
- Zhang, W.; Shen, X.Y.; Zhang, W.W.; Chen, H.; Xu, W.P.; Wei, W. The Effects of Di 2-Ethyl Hexyl Phthalate (DEHP) on Cellular Lipid Accumulation in HepG2 Cells and Its Potential Mechanisms in the Molecular Level. Toxicol. Mech. Methods 2017, 27, 245–252.
- Huff, M.; da Silveira, W.A.; Carnevali, O.; Renaud, L.; Hardiman, G. Systems Analysis of the Liver Transcriptome in Adult Male Zebrafish Exposed to the Plasticizer (2-Ethylhexyl) Phthalate (DEHP). Sci. Rep. 2018, 8, 2118.
- Radke, E.G.; Galizia, A.; Thayer, K.A.; Cooper, G.S. Phthalate Exposure and Metabolic Effects: A Systematic Review of the Human Epidemiological Evidence. Environ. Int. 2019, 132, 104768.
- Sun, Q.; Cornelis, M.C.; Townsend, M.K.; Tobias, D.K.; Eliassen, A.H.; Franke, A.A.; Hauser, R.; Hu, F.B. Association of Urinary Concentrations of Bisphenol A and Phthalate Metabolites with Risk of Type 2 Diabetes: A Prospective Investigation in the Nurses’ Health Study (NHS) and NHSII Cohorts. Environ. Health Perspect. 2014, 122, 616–623.
- Badamasi, I.; Odong, R.; Masembe, C. Threats Posed by Xenoestrogenic Chemicals to the Aquatic Ecosystem, Fish Reproduction and Humans: A Review. Afr. J. Aquat. Sci. 2020, 45, 243–258.
- Laura N. Vandenberg; Theo Colborn; Tyrone B. Hayes; Jerrold J. Heindel; David R. Jacobs; Duk-Hee Lee; John Peterson Myers; Toshi Shioda; Ana M. Soto; Frederick S. Vom Saal; et al.Wade V. WelshonsR. Thomas Zoeller Regulatory decisions on endocrine disrupting chemicals should be based on the principles of endocrinology. Reproductive Toxicology 2013, 38, 1-15, 10.1016/j.reprotox.2013.02.002.
- Christopher Kassotis; Laura N Vandenberg; Barbara A Demeneix; Miquel Porta; Remy Slama; Leonardo Trasande; Endocrine-disrupting chemicals: economic, regulatory, and policy implications. The Lancet Diabetes & Endocrinology 2020, 8, 719-730, 10.1016/s2213-8587(20)30128-5.
- Derek V. Henley; Kenneth Korach; Endocrine-Disrupting Chemicals Use Distinct Mechanisms of Action to Modulate Endocrine System Function. Endocrinology 2006, 147, s25-s32, 10.1210/en.2005-1117.
- Bhasker, C.R.; Hardiman, G. Advances in Pharmacogenomics Technologies. Pharmacogenomics 2010, 11, 481–485.
- Miracle, A.L.; Ankley, G.T. Ecotoxicogenomics: Linkages between Exposure and Effects in Assessing Risks of Aquatic Contaminants to Fish. Reprod. Toxicol. 2005, 19, 321–326.
- Martín-Díaz, M.L.; DelValls, T.A.; Riba, I.; Blasco, J. Integrative Sediment Quality Assessment Using a Biomarker Approach: Review of 3 Years of Field Research. Cell. Biol. Toxicol. 2008, 24, 513–526.
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev. 2009, 30, 293–342.
- Grün, F.; Blumberg, B. Endocrine Disrupters as Obesogens. Mol. Cell. Endocrinol. 2009, 304, 19–29.
- Baker, M.E.; Sprague, L.J.; Ribecco, C.; Ruggeri, B.; Lekmine, N.; Ludka, C.; Wick, I.; Soverchia, L.; Ubaldi, M.; Šášik, R.; et al. Application of a Targeted Endocrine Q-PCR Panel to Monitor the Effects of Pollution in Southern California Flatfish. Endocr. Disruptors 2014, 2, e969598.
- Baker, M.E.; Vidal-Dorsch, D.E.; Ribecco, C.; Sprague, L.J.; Angert, M.; Lekmine, N.; Ludka, C.; Martella, A.; Ricciardelli, E.; Bay, S.M.; et al. Molecular Analysis of Endocrine Disruption in Hornyhead Turbot at Wastewater Outfalls in Southern California Using a Second Generation Multi-Species Microarray. PLoS ONE 2013, 8, e75553.
- Hardiman, G. Microarray Technology—Advances, Applications, Future Prospects. Pharmacogenomics 2007, 8, 1639–1642.
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63.
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Liguori, M.J. Comparison of RNA-Seq and Microarray Gene Expression Platforms for the Toxicogenomic Evaluation of Liver From Short-Term Rat Toxicity Studies. Front. Genet. 2019, 9, 636.
- Casneuf, T.; Van de Peer, Y.; Huber, W. In Situ Analysis of Cross-Hybridisation on Microarrays and the Inference of Expression Correlation. BMC Bioinform. 2007, 8, 461.
- Chen, C.; Grennan, K.; Badner, J.; Zhang, D.; Gershon, E.; Jin, L.; Liu, C. Removing Batch Effects in Analysis of Expression Microarray Data: An Evaluation of Six Batch Adjustment Methods. PLoS ONE 2011, 6, e17238.
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969.
- Microarray Innovations: Technology and Experimentation. Available online: https://www.routledge.com/Microarray-Innovations-Technology-and-Experimentation/Hardiman/p/book/9780367385811 (accessed on 20 July 2021).
- Rouse, R.J.D.; Field, K.; Lapira, J.; Lee, A.; Wick, I.; Eckhardt, C.; Bhasker, C.R.; Soverchia, L.; Hardiman, G. Development and Application of a Microarray Meter Tool to Optimize Microarray Experiments. BMC Res. Notes 2008, 1, 45.
- Wick, I.; Hardiman, G. Biochip Platforms as Functional Genomics Tools for Drug Discovery. Curr. Opin. Drug Discov. Dev. 2005, 8, 347–354.
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PLoS ONE 2014, 9, e78644.
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem Cell Transcriptome Profiling via Massive-Scale MRNA Sequencing. Nat. Methods 2008, 5, 613–619.
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science 2008, 320, 1344–1349.
- da Silveira, W.A.; Hazard, E.S.; Chung, D.; Hardiman, G. Molecular Profiling of RNA Tumors Using High-Throughput RNA Sequencing: From Raw Data to Systems Level Analyses. Methods Mol. Biol. 2019, 1908, 185–204.
- Davis-Turak, J.; Courtney, S.M.; Hazard, E.S.; Glen, W.B.; da Silveira, W.A.; Wesselman, T.; Harbin, L.P.; Wolf, B.J.; Chung, D.; Hardiman, G. Genomics Pipelines and Data Integration: Challenges and Opportunities in the Research Setting. Expert Rev. Mol. Diagn. 2017, 17, 225–237.
- Rigden, D.J.; Fernández, X.M. The 2021 Nucleic Acids Research Database Issue and the Online Molecular Biology Database Collection. Nucleic Acids Res. 2021, 49, D1–D9.
- Lawler, M.; Siu, L.L.; Rehm, H.L.; Chanock, S.J.; Alterovitz, G.; Burn, J.; Calvo, F.; Lacombe, D.; Teh, B.T.; North, K.N.; et al. All the World’s a Stage: Facilitating Discovery Science and Improved Cancer Care through the Global Alliance for Genomics and Health. Cancer Discov. 2015, 5, 1133–1136.
- Leet, J.K.; Richter, C.A.; Cornman, R.S.; Berninger, J.P.; Bhandari, R.K.; Nicks, D.K.; Zajicek, J.L.; Blazer, V.S.; Tillitt, D.E. Effects of Early Life Stage Exposure of Largemouth Bass to Atrazine or a Model Estrogen (17α-Ethinylestradiol). PeerJ 2020, 8, e9614.
- Wang, J.; Zhou, J.; Yang, Q.; Wang, W.; Liu, Q.; Liu, W.; Liu, S. Effects of 17 α-Methyltestosterone on the Transcriptome, Gonadal Histology and Sex Steroid Hormones in Pseudorasbora Parva. Theriogenology 2020, 155, 88–97.
- Renaud, L.; Agarwal, N.; Richards, D.J.; Falcinelli, S.; Hazard, E.S.; Carnevali, O.; Hyde, J.; Hardiman, G. Transcriptomic Analysis of Short-Term 17α-Ethynylestradiol Exposure in Two Californian Sentinel Fish Species Sardine (Sardinops sagax) and Mackerel (Scomber japonicus). Environ. Pollut. 2019, 244, 926–937.
- Bertucci, A.; Pierron, F.; Gourves, P.-Y.; Klopp, C.; Lagarde, G.; Pereto, C.; Dufour, V.; Gonzalez, P.; Coynel, A.; Budzinski, H.; et al. Whole-Transcriptome Response to Wastewater Treatment Plant and Stormwater Effluents in the Asian Clam, Corbicula fluminea. Ecotoxicol. Environ. Saf. 2018, 165, 96–106.
- Legrand, E.; Forget-Leray, J.; Duflot, A.; Olivier, S.; Thomé, J.-P.; Danger, J.-M.; Boulangé-Lecomte, C. Transcriptome Analysis of the Copepod Eurytemora Affinis upon Exposure to Endocrine Disruptor Pesticides: Focus on Reproduction and Development. Aquat. Toxicol. 2016, 176, 64–75.
- Guo, J.; Mo, J.; Qi, Q.; Peng, J.; Qi, G.; Kanerva, M.; Iwata, H.; Li, Q. Prediction of Adverse Effects of Effluents Containing Phenolic Compounds in the Ba River on the Ovary of Fish (Hemiculter leucisculus) Using Transcriptomic and Metabolomic Analyses. Sci. Total Environ. 2021, 801, 149554.
- Garcia-Reyero, N. Are Adverse Outcome Pathways Here to Stay? Environ. Sci. Technol. 2015, 49, 3–9.
- Perkins, E.J.; Antczak, P.; Burgoon, L.; Falciani, F.; Garcia-Reyero, N.; Gutsell, S.; Hodges, G.; Kienzler, A.; Knapen, D.; McBride, M.; et al. Adverse Outcome Pathways for Regulatory Applications: Examination of Four Case Studies with Different Degrees of Completeness and Scientific Confidence. Toxicol. Sci. 2015, 148, 14–25.
- Sushila Arya; Alok Kumar Dwivedi; Luis Alvarado; Sanja Kupesic-Plavsic; Exposure of U.S. population to endocrine disruptive chemicals (Parabens, Benzophenone-3, Bisphenol-A and Triclosan) and their associations with female infertility. Environmental Pollution 2020, 265, 114763, 10.1016/j.envpol.2020.114763.
- Villeneuve, D.L.; Crump, D.; Garcia-Reyero, N.; Hecker, M.; Hutchinson, T.H.; LaLone, C.A.; Landesmann, B.; Lettieri, T.; Munn, S.; Nepelska, M.; et al. Adverse Outcome Pathway (AOP) Development I: Strategies and Principles. Toxicol. Sci. 2014, 142, 312–320.
- Yoshinobu Igarashi; Noriyuki Nakatsu; Tomoya Yamashita; Atsushi Ono; Yasuo Ohno; Tetsuro Urushidani; Hiroshi Yamada; Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Research 2014, 43, D921-D927, 10.1093/nar/gku955.
- Naoki Kiyosawa; Sunao Manabe; Atsushi Sanbuissho; Takashi Yamoto; Gene set-level network analysis using a toxicogenomics database. Genomics 2010, 96, 39-49, 10.1016/j.ygeno.2010.03.014.
- Naoki Kiyosawa; Sunao Manabe; Atsushi Sanbuissho; Takashi Yamoto; Gene set-level network analysis using a toxicogenomics database. Genomics 2010, 96, 39-49, 10.1016/j.ygeno.2010.03.014.
More