Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Nicolai Bjødstrup Palstrøm.
The human plasma proteome is an immensely complex mixture of proteins. Astonishingly, only 730 proteins are known to be secreted into the blood whereas the vast majority of plasma proteins are present as a result of leakage into the blood due to tissue degradation and damage, i.e., the plasma proteome potentially comprises of proteins that were derived from all tissues.
- plasma proteomics
- cardiovascular disease
1. Introduction
In recent years, there has been a rise in the use of proteomic approaches for the study of bodily fluids to uncover disease mechanisms that are associated with the cardiovascular system and to improve diagnosis, prognosis, and monitoring in cardiovascular diseases (CVD). As CVDs remain a prominent cause of morbidity and death, interest in establishing CVD-related biomarkers has steadily grown [1]. Recognizing risk factors such as tobacco usage, diabetes, hypertension, and hyperlipidemia in individuals is currently the dominant method of identifying those who are at risk of developing CVDs. [2]. Currently, some plasma proteins are routinely investigated for decision making in cardiovascular medicine. Examples of the most prominent ones are creatine kinase and troponins, and natriuretic peptides for the diagnosis of myocardial infarcts and heart failure. In addition, lipoproteins are promising for the stratification of cardiovascular risk. However, it is common for these markers that each of them is limited in their diagnostic or predictive accuracy. Therefore, high-performance biomarkers for CVD stratification, diagnosis of acute myocardial infarct (AMI), heart failure (HF), and other cardiovascular conditions, such as abdominal aortic aneurysms are urgently needed. Biomarkers for cardiovascular diagnostics should ideally have a high sensitivity, possess high tissue specificity, and the measured concentration should be proportional to the severity of the specific CVD [3]. The clinical cohorts from which biomarkers for CVD is derived from should be clearly defined in terms of the study population regarding e.g., sex, ethnicity, age, preexisting conditions, and other factors that are known to influence CVDs and also should be accounted for during statistical analysis [4].
The plasma proteome contains thousands of proteins and the general assumption is that most of them remain unexplored for their relation to multiple diseases including CVDs. Therefore, a systematic exploration of all proteins that are present in plasma holds great potential for displaying novel biomarkers for diagnostic, prognostic, and monitoring purposes. Historically, barriers for plasma proteomics have mainly been due to technical limitations, for example, a lack of sensitivity and specificity of the applied analytical method as well as limited possibilities for multiplexing and a low sample throughput. Complementing the information that was obtained from traditional risk factors with measurements of the relevant biomarkers in body fluids, the individualized risk stratification and diagnosis of CVD may lead the way for improved patient classification [5]. Proteomics covers a variety of analysis principles spanning both explorative and unbiased methods. For example, 2-gel electrophoresis (2DE) coupled to matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF) as well as two-dimension liquid chromatography in combination with tandem mass spectrometry are often applied in discovery proteomics. On the other hand, affinity-based methods such as multiplex immunoassays and aptamer-based assays target arrays of specific proteins [6,7][6][7].
2. Recent Developments in Assaying the Human Plasma Proteome by Proteomics Methods
The human plasma proteome displays an extreme complexity in terms of function and protein abundance for specific proteins. This complexity transforms into an extreme analytical complexity when it comes to the proteomic analysis of individual proteins at low abundance in the presence of very high abundant proteins. The predominance of high abundant so-called classical plasma proteins complicates the detection of the lower abundant ones by compromising the dynamic range of the analytical methods in question; in particular, the traditionally used methods for plasma proteome analysis, such gel-based electrophoretic methods (1- and 2-DE) and LC methods combined with MALDI-TOF-MS and MSMS analysis. These methods, introduced in the late 1990s and early 2000s, were limited to the detection of most abundant proteins in plasma. Since then several attempts to counteract the challenges of analyzing low-abundant proteins in plasma have been investigated throughout the years by the introduction of a tremendous number novel methods for sample preparation of human plasma, data acquisition methods for mass spectrometry-based plasma proteomics, multiplex affinity-based assays that rely on binding of specific proteins to short oligomers, or to antibodies enabling the simultaneous targeted measurement of 100 to thousands of proteins.
In the remaining sections, we will introduce the most recent and commonly used proteomic methods that are applied to plasma proteomics in cardiovascular medicine. This includes, mass spectrometry, aptamer-based microarray (SomaScan), and immunoaffinity assays (Olink). The principle of these methods are summarized in Figure 1.
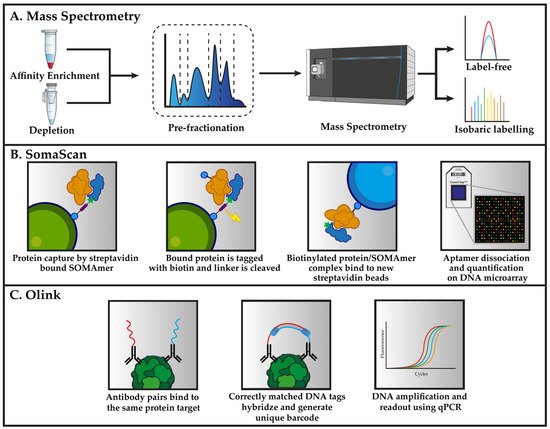
Figure 1. Workflows for the three primary analytical techniques that were deployed for proteomic analysis of plasma. (A) Mass spectrometry-based methods for the analysis of plasma typically relies on either depletion or enrichment to detect low-abundant proteins. Pre-fractionation prior to proteomic analysis is an optional step that separates the peptides in one dimension, thereby dividing the sample into a smaller fraction for later analysis. The quantification of proteins for explorative biomarker discovery is performed either label-free or using isobaric labelling reagents. (B) The SomaScan assay uses SOMAmers (blue) that are synthesized with a fluorophore, photocleavable linker as well as biotin, which is used to immobilize the SOMAmers to streptavidin beads. SOMAmer are able to capture proteins (orange) from solution followed by biotinylating of the captured proteins. The photocleavable linker is destroyed using external UV light to release the SOMAmer/protein complex back into the solution. Biotin-tagged proteins are recaptured on secondary streptavidin beads. SOMAmer dissociation is caused by denaturing of the captured protein, thereby allowing the SOMAmer reagents to hybridize to complementary sequences on a microarray chip. The abundance of each protein is derived from the amount of fluorescent intensity that is detected from each fluorophore. (C) Antibodies with the specificity for the same protein (green) are brought into close enough proximity that the attached DNA can hybridize and extend in the presence of a DNA polymerase. This process forms unique DNA barcodes for each protein in the assay, which can then be amplified and used for quantification using qPCR.
2.1. Mass Spectrometry-Based Plasma Proteomics
2.1.1. Preanalytical Steps in Mass Spectrometry-Based Plasma Proteomics
As mentioned above, the complexity of the plasma proteome hinders the detection of lower abundant proteins by mass spectrometry-based proteomics. Therefore, much effort has been devoted to the development of methods, such as selective precipitation using acetonitrile [35][8], protein equalization using combinatorial peptide ligand libraries (ProteoMiner) [36][9], or immunodepletion for the removal of up to the 50 of the most abundant proteins in human plasma [37,38][10][11] prior to mass spectrometry analysis. Currently, the commonly used methods for mass spectrometry-based plasma proteomics are the two latter ones. Despite the fact that immunodepletion methods and the protein equalization methods [37,38][10][11] reduce the signals from high abundant proteins in the mass spectrometry analysis, they do not allow the detection of low abundant plasma protein by mass spectrometry. Recently, a method that was based on the affinity capture of low-abundant plasma proteins using small-molecule affinity-based probes was developed [39][12]. A total of four different small-molecule affinity-based probes that were based on agarose-immobilized p-aminobenzamidine (ABA), 8-amino-hexyl-ATP (ATP), 8-amino-hexyl-cAMP (cAMP), or O-phospho-L-tyrosine (pTYR) were explored for their capability to enrich lower abundant proteins while also removing high-abundant proteins. They demonstrated excellent removal of high abundant plasma proteins and selective enrichment of low abundant plasma proteins as compared with the immuno-depletion and protein equalization and outperformed the other methods. Compared to undepleted plasma, a more than an 80% increase in protein identification was obtained based on these methods [39][12]. This method was applied for the proteomics detection of low-abundant proteins that were originally identified as biomarkers for AMI, which, for a large part, turned out to be confounded by heparin administration [40][13], as well as in a study that identified low abundant protein biomarkers for cardiogenic shock [41][14]. In addition to the challenge of detecting low abundant proteins, the introduction of contaminants during sample handling and the use of detergents is also something to consider during sample preparation as it can negatively influence the quality of the data that are generated [42][15]. Other issues regarding incomplete proteolysis, inadequate protein solubilization, and lack of a desalting step are important factors that can have an impact on experiments [43][16].
2.1.2. MS-Based Proteomics: LC-MSMS Analysis
The preferred method for high throughput proteomic analysis of human plasma has, since the early 2000s, been liquid chromatography combined with mass spectrometry (LC-MS) [44,45][17][18]. Generically, a mass spectrometer measures mass–charge ratios of ionized analytes (e.g., peptides or proteins) within a gas phase. Although many diverse types of mass spectrometers exist, the workflows for plasma proteomics are typically based on the depletion of high-abundant proteins or enrichment of low abundant proteins. This occurs via the proteolytic cleavage of the proteins into well-defined peptides by restriction enzymes, typically trypsin, followed by chromatographic separation using nano-flow LC and mass spectrometric detection that measures both the accurate mass and the amino acid sequence of the tryptic peptides. The main issue with LC-MS applied to plasma proteome analysis is that it is not possible with this method to measure across the entire dynamic range of plasma, and it, therefore, requires additional analytical steps [10][19]. The large majority of mass spectrometry-based analyses on plasma is performed with what is known as a bottom-up proteomic approach [46][20]. In bottom-up proteomics (sometime referred to as shotgun proteomics), the proteins are proteolytically digested and are then chromatographically separated (in one or several dimensions) and the analysis is performed on the resulting peptides. In principle, two complementary approaches are employed for peptide measurements using MS; untargeted MS and targeted MS.
For un-targeted MS, two methods for data-acquisition are applied for MS-based plasma proteomics: data-dependent acquisition (DDA) and data-independent acquisition (DIA). The most commonly used mode of data-acquisition in mass spectrometry is DDA in which precursor ions whose intensity exceed a pre-defined threshold in a survey scan (MS1) are automatically selected for fragmentation. This (semi) stochastic way of selecting precursor ions is inherently biased toward the most abundant ions and also leads to the selection of different subsets of ions in subsequent analytical runs leading to a higher degree of missing values and less reproducibility between runs [47][21]. Not surprisingly, this also implies that the biologically relevant precursor ions that fail to meet the pre-defined threshold or are co-eluted with more abundant ions will not necessarily be selected for fragmentation [48][22]. A way to reduce the influence of high abundant ions is the use of dynamic exclusion which prevents the repeated acquisition of precursor ions and can have a noticeable effect on the number of the identified proteins. The downside of bottom-up proteomics is that the identification of proteins is limited to the canonical amino acid sequences and, less reliably, the isoforms of said canonical proteins, unless their amino acid sequences are present in the database that is being used for searching the data. The identification of different proteoforms is hindered largely due to the proteolytic digestion which is an unavoidable aspect of bottom-up proteomics. On the other hand, the nature of PTM can be localized by DDA acquisition, thereby enabling the investigation of the relevance of PTMs in cardiovascular diseases [49][23] as long as the PTM is located in a canonical protein sequence on sequencing that is present in the database that is being applied for searching the raw data. The processing of raw data from DDA have been made easily available with both commercial and open-source pipelines for the identification of protein identities [50,51,52][24][25][26]. In DDA analysis, protein identities are inferred based on sequencing of the proteolytically digested peptide by searching the derived peptide sequence against a protein sequence database, such as UniProt [53][27].
With DIA, some of the shortcomings of DDA, such as stochastic and irreproducible ion selection, is alleviated by removing the direct dependency on ion intensity for ion fragmentation. A typical implementation of DIA is sequential window acquisition of all theoretical mass spectra (SWATH-MS). In its first implementation by Gillet et al. [54][28], a quadrupole-Time-of-Flight (q-TOF) mass spectrometer was used to cycle through 32 consecutive, overlapping (1 m/z) precursor isolation windows with a width of 25 Da (so called swaths) for systematic and unbiased fragmentation of precursor ions that fell within the predetermined isolation windows. While data acquisition with DIA is fairly simple, the analysis of DIA data is still currently a challenge due to the multiplexed MS2 spectra that are generated due to the co-fragmentation of co-eluting precursor ions. Originally, DIA data was analyzed using similar database search engines that were used in DDA data analysis. However, Gillet et al. also devised a novel data analysis workflow in which prior knowledge of previously observed peptides that were contained in spectral libraries was used to search the generated data. Generally, DIA has several advantages in relation to DDA analysis, such as higher reproducibility and retrospective querying [55,56][29][30].
With targeted MS, stable isotope peptides are used as reference points to provide absolute quantification of the peptides that are present in the sample. Targeted MS, also referred to as multiple reaction monitoring (MRM) is predominantly used for studies where a relatively small number of preselected proteins are measured. For example, for the validation of protein biomarkers in large sample cohort that was independent of the cohort that was analyzed, a more global untargeted proteomic strategy was used to initially discover them. While MRM is usually performed with triple-quadrupole instruments, another implementation is parallel reaction monitoring (PRM) in which the high-resolution Orbitrap or alternatively a time-of-flight (TOF) is used as an MS2 analyzer [57,58][31][32]. The drawback of targeted MRM analysis is the need for prior knowledge of the peptides of interest with regards to the transitions of the fragment ions [59][33]. This drawback is, however, outweighed by the fact that MRM provides a more sensitive and also an absolute quantification which enables a comparison of results across studies from different laboratories. This limitation is alleviated with PRM in which prior knowledge of the target transitions is unneeded due to the simultaneous monitoring of product ions of a targeted peptide with high resolution. Both SRM and PRM have demonstrated comparable sensitivities, repeatability, and dynamic range for targeted quantification [60,61][34][35]. A classic example in cardiovascular research where protein biomarkers are initially identified by untargeted MS and validated by targeted MS include the identification and validation of vinculin as plasma biomarker for acute coronary syndrome [62][36].
2.1.3. Quantitative MS-Based Proteomics
One of the key important prerequisites for success in finding new, CVD-specific biomarkers are that the methods that are used are quantitative, that these methods have high analytical precision, and that there is a sufficient number of subjects in the cohort being studied, i.e., that the study is adequately powered. The quantification of proteins in DDA discovery studies is performed in two ways: as label-free quantification, or with the use of protein-labelling reagents, so-called isobaric tags. Label-free quantification extracts the MS1 peptide precursor ion chromatogram and uses either the intensity of the highest point or integrates the peak area over the chromatographic time scale [63][37] as a quantitative measure. Another way of label-free-quantification is spectral counting in which the number peptide spectrum matches (PSMs) from identified unique peptides from a given protein are summed as the number of PSMs that have been shown to correlate with the protein quantity [64][38]. Label-free quantification methods often leads to a higher degree of missing values in the quantification step due to the stochastic sampling characteristic of DDA analyses meaning that the same proteins are not identified and quantified across different samples [65][39]. A process that is primarily known as match-between runs (available in free-to-use software, such as MaxQuant) in which protein identities and quantification can be transferred between files based on MS1 scans, leads to significant reduction in the number of missing values [66][40].
With quantification using isobaric tags, isobaric amine-reactive tags are covalently coupled to the reactive amino groups of lysine and the peptide N-terminus [67][41]. Different tagging methods are commercially available [68,69][42][43] and allows the multiplexing of up to 18 and subsequently analyze them in one single MS experiment [70][44]. This analytical setup is increasingly being applied to large-scale proteomics studies allowing for highly complex study designs [71,72][45][46] for the analysis of a large number of patient samples. In a multiplex isobaric-tagging experiment, each individual sample is digested and peptides are labeled/tagged with a unique label/tag and pooled in equimolar proportions. The peptides tagged with labeling reagent are fragmented with high-energy collision-induced dissociation (HCD) during analysis and the mass reporter ions are released [73][47]. The intensities of each different mass reporter ion are then representative for the relative concentration of the measured peptides in each sample. The use of isobaric-tagging enables high-throughput analysis which reduces the instrument time, variation between runs, and the extent of missing values [74][48].
2.2. Affinity-Based Proteomics Methods
The pace by which new developments of affinity-based techniques that are used in sample processing for increasing the measurable dynamic range, have increased rapidly. Typically, depletion methods as well as substantial fractionation have been employed to lessen the effect of high abundant proteins in mass spectrometry analysis.
In recent years, several commercially available affinity-based platforms promising high-throughput analysis of plasma with high sensitivity and the capacity to analyze several hundred to thousands of proteins in a multiplex manner and have been developed and applied in numerous CVD biomarker discovery studies. The plasma proteome has an enormous dynamic range and variability, including splice variants, cleavage products, and posttranslational modifications. Antibody-based techniques have, so far, predominated, but recent developments in affinity-based techniques such as the SomaScan aptamer assays (SomaLogic), proximity extension assays (Olink), and microbead-based multiplex immunoassay (xMAP), also promise the multiplexed measurement of proteins in a scalable manner.
2.2.1. Aptamer Microarrays (SomaScan)
Methods that use DNA or RNA scaffolds as binding reagents have emerged during recent years and one of these is the SomaScan assay, which uses single-stranded DNA-based protein affinity reagents, termed SOMAmer (Slow Off-rate Modified Aptamer). Publications which use the SomaScan-platform have increased over the years, particularly in the search for novel CVD plasma biomarkers. The attraction is easily understood as each iteration of the platform has increased the number of proteins that can be measured in a single run, with the latest iteration of the platform offering ≈ 7000 protein measurements. The platform utilizes aptamers (short oligonucleotides) with binding affinities to the epitopes of the selected proteins, which are multiplexed allowing the simultaneous quantification of multiple proteins at the same time. The SomaScan assay exploits the versatility of oligonucleotides in their capability of binding specific proteins as well as being detectable by DNA detection methods. Modified nucleotides that are combined with an artificial iterative selection process of appropriate aptamers (SELEX) enables the creation of reagents with specificity for target proteins [75][49]. Overall, the SomaScan assay targets a large subset of the human plasma proteome but claims to overcome classical dynamic range limitations that are observed with MS methods. While the SomaScan assay is best known for high throughput screening of clinical samples in typical biofluids, e.g., serum, plasma, and cerebrospinal fluid, others have also used the assay to study exosomes and cell extracts [76,77,78,79][50][51][52][53]. Moreover, SomaScan assay was recently applied to the proteomic analysis of plasma in relation to heart failure [80][54] and acute myocardial infarction (AMI) [81][55].
2.2.2. Proximity Extension Assays (Olink)
Another recent development in multiplex affinity-based plasma proteomics is the proximity extension assay (PEA) technology that was commercialized by Olink Proteomics AB (Uppsala, Sweden). Both exploratory 384-plex kits that were based on a next-generation sequencing (NGS) platform or more targeted solutions using 48-96-plex kits that were based on quantitative real-time PCR (qPCR) platform are available with focus on different groups of proteins (e.g., inflammatory- or cardiovascular-related proteins). The Olink assay uses pairs of antibodies for the dual recognition of target proteins, thereby increasing specificity. Unique DNA oligonucleotides are attached to each antibody and will hybridize only when in close proximity to a matched antibody. The DNA-polymerase-mediated extension of the hybridized oligonucleotides forms a unique barcode-sequence that is specific for each protein. The created sequence can then be quantified using either qPCR or NGS (depending on the kits used) as the initial concentration of target proteins is proportional to the quantity of generated sequences through the polymerase reaction. The Olink platform has recently been applied for investigating circulating biomarkers for heart failure [82][56] and coronary heart disease [83][57].
2.2.3. Microbead-Based Multiplex Immunoassay (xMAP)
The Luminex xMAP (Multi-Analyte profiling) technology uses different sets of microspheres in either a magnetic or non-magnetic bead format. Microsphere sets are internally dyed with two spectrally different fluorophores in different concentrations which creates unique spectral signatures for each microsphere set. Each microsphere set can be coated with various capture molecules, facilitating the capture of up to 100 analytes at the same time. Excitation with a dual laser system enables differentiating between the different microsphere sets and parallel measurement of the fluorescent reporter molecule, which has been captured with the assay. The xMAP platform has recently been used to investigate biomarkers for different cardiovascular diseases [84][58] and another study investigating atrial fibrillation [85][59].
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492.
- Yusuf, S.; Joseph, P.; Rangarajan, S.; Islam, S.; Mente, A.; Hystad, P.; Brauer, M.; Kutty, V.R.; Gupta, R.; Wielgosz, A.; et al. Modifiable risk factors, cardiovascular disease, and mortality in 155 722 individuals from 21 high-income, middle-income, and low-income countries (PURE): A prospective cohort study. Lancet 2020, 395, 795–808.
- Dhingra, R.; Vasan, R.S. Biomarkers in cardiovascular disease: Statistical assessment and section on key novel heart failure biomarkers. Trends Cardiovasc. Med. 2017, 27, 123–133.
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 2021, 16, 3737–3760.
- Dogan, M.V.; Beach, S.R.H.; Simons, R.L.; Lendasse, A.; Penaluna, B.; Philibert, R.A. Blood-Based Biomarkers for Predicting the Risk for Five-Year Incident Coronary Heart Disease in the Framingham Heart Study via Machine Learning. Genes 2018, 9, 641.
- Hedl, T.J.; Gil, R.S.; Cheng, F.; Rayner, S.; Davidson, J.M.; De Luca, A.; Villalva, M.D.; Ecroyd, H.; Walker, A.K.; Lee, A. Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front. Neurosci. 2019, 13, 548.
- Khalilpour, A.; Kilic, T.; Alvarez, M.M.; Yazdi, I.K. Proteomic-based biomarker discovery for development of next generation diagnostics. Appl. Microbiol. Biotechnol. 2017, 101, 475–491.
- Mostovenko, E.; Scott, H.C.; Klychnikov, O.; Dalebout, H.; Deelder, A.M.; Palmblad, M. Protein Fractionation for Quantitative Plasma Proteomics by Semi-Selective Precipitation. J. Proteom. Bioinform. 2012, 5, 217–221.
- Boschetti, E.; Righetti, P.G. The ProteoMiner in the proteomic arena: A non-depleting tool for discovering low-abundance species. J. Proteom. 2008, 71, 255–264.
- Shi, T.; Zhou, J.-Y.; Gritsenko, M.A.; Hossain, M.; Camp, D.G.; Smith, R.D.; Qian, W.-J. IgY14 and SuperMix immunoaffinity separations coupled with liquid chromatography–mass spectrometry for human plasma proteomics biomarker discovery. Methods 2012, 56, 246–253.
- Beer, L.A.; Ky, B.; Barnhart, K.T.; Speicher, D.W. In-Depth, Reproducible Analysis of Human Plasma Using IgY 14 and SuperMix Immunodepletion. Methods Mol. Biol. 2017, 1619, 81–101.
- Palstrøm, N.B.; Rasmussen, L.M.; Beck, H.C. Affinity Capture Enrichment versus Affinity Depletion: A Comparison of Strategies for Increasing Coverage of Low-Abundant Human Plasma Proteins. Int. J. Mol. Sci. 2020, 21, 5903.
- Beck, H.C.; Jensen, L.O.; Gils, C.; Ilondo, A.M.M.; Frydland, M.; Hassager, C.; Møller-Helgestad, O.K.; Møller, J.E.; Rasmussen, L.M. Proteomic Discovery and Validation of the Confounding Effect of Heparin Administration on the Analysis of Candidate Cardiovascular Biomarkers. Clin. Chem. 2018, 64, 1474–1484.
- Debrabant, B.; Halekoh, U.; Soerensen, M.; Møller, J.E.; Hassager, C.; Frydland, M.; Palstrøm, N.; Hjelmborg, J.; Beck, H.C.; Rasmussen, L.M. STEMI, Cardiogenic Shock, and Mortality in Patients Admitted for Acute Angiography: Associations and Predictions from Plasma Proteome Data. Shock 2021, 55, 41–47.
- Keller, B.O.; Sui, J.; Young, A.B.; Whittal, R.M. Interferences and contaminants encountered in modern mass spectrometry. Anal. Chim. Acta 2008, 627, 71–81.
- DuPree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of this Field. Proteomes 2020, 8, 14.
- Woods, A.G.; Sokolowska, I.; Wetie, A.G.N.; Channaveerappa, D.; Dupree, E.J.; Jayathirtha, M.; Aslebagh, R.; Wormwood, K.L.; Darie, C.C. Mass Spectrometry for Proteomics-Based Investigation. Adv. Exp. Med. Biol. 2019, 1140, 1–26.
- Gillet, L.C.; Leitner, A.; Aebersold, R. Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. Annu. Rev. Anal. Chem. 2016, 9, 449–472.
- Kaur, G.; Poljak, A.; Ali, S.A.; Zhong, L.; Raftery, M.J.; Sachdev, P. Extending the Depth of Human Plasma Proteome Coverage Using Simple Fractionation Techniques. J. Proteome Res. 2021, 20, 1261–1279.
- Baker, E.S.; Liu, T.; Petyuk, A.V.; Burnum-Johnson, E.K.; Ibrahim, Y.M.; Anderson, A.G.; Smith, R.D. Mass spectrometry for translational proteomics: Progress and clinical implications. Genome Med. 2012, 4, 63.
- Wang, J.; Tucholska, M.; Knight, J.D.; Lambert, J.-P.; Tate, S.; Larsen, B.; Gingras, A.-C.; Bandeira, N. MSPLIT-DIA: Sensitive peptide identification for data-independent acquisition. Nat. Methods 2015, 12, 1106–1108.
- Hilaire, P.B.S.; Rousseau, K.; Seyer, A.; Dechaumet, S.; Damont, A.; Junot, C.; Fenaille, F. Comparative Evaluation of Data Dependent and Data Independent Acquisition Workflows Implemented on an Orbitrap Fusion for Untargeted Metabolomics. Metabolites 2020, 10, 158.
- Smith, L.M.; Kelleher, N.L. Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107.
- Orsburn, B. Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes 2021, 9, 15.
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372.
- Röst, H.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748.
- The UniProt Consortium. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169.
- Gillet, L.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11, O111.016717.
- Barkovits, K.; Pacharra, S.; Pfeiffer, K.; Steinbach, S.; Eisenacher, M.; Marcus, K.; Uszkoreit, J. Reproducibility, Specificity and Accuracy of Relative Quantification Using Spectral Library-based Data-independent Acquisition. Mol. Cell. Proteom. 2020, 19, 181–197.
- Rosenberger, G.; Liu, Y.; Röst, H.L.; Ludwig, C.; Buil, A.; Bensimon, A.; Soste, M.; Spector, T.D.; Dermitzakis, E.T.; Collins, B.C.; et al. Inference and quantification of peptidoforms in large sample cohorts by SWATH-MS. Nat. Biotechnol. 2017, 35, 781–788.
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581.
- Michalski, A.; Damoc, E.; Hauschild, J.-P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass Spectrometry-based Proteomics Using Q Exactive, a High-performance Benchtop Quadrupole Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2011, 10, M111.011015.
- Zybailov, B.; Mosley, A.L.; Sardiu, M.E.; Coleman, M.K.; Florens, L.; Washburn, M.P. Statistical Analysis of Membrane Proteome Expression Changes in Saccharomyces cerevisiae. J. Proteome Res. 2006, 5, 2339–2347.
- Schiffmann, C.; Hansen, R.; Baumann, S.; Kublik, A.; Nielsen, P.H.; Adrian, L.; von Bergen, M.; Jehmlich, N.; Seifert, J. Comparison of targeted peptide quantification assays for reductive dehalogenases by selective reaction monitoring (SRM) and precursor reaction monitoring (PRM). Anal. Bioanal. Chem. 2014, 406, 283–291.
- Ronsein, G.E.; Pamir, N.; von Haller, P.D.; Kim, D.S.; Oda, M.N.; Jarvik, G.P.; Vaisar, T.; Heinecke, J.W. Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J. Proteom. 2015, 113, 388–399.
- Kristensen, L.P.; Larsen, M.R.; Mickley, H.; Saaby, L.; Diederichsen, A.C.; Lambrechtsen, J.; Rasmussen, L.M.; Overgaard, M. Plasma proteome profiling of atherosclerotic disease manifestations reveals elevated levels of the cytoskeletal protein vinculin. J. Proteom. 2014, 101, 141–153.
- Lindemann, C.; Thomanek, N.; Hundt, F.; Lerari, T.; Meyer, H.E.; Wolters, D.; Marcus, K. Strategies in relative and absolute quantitative mass spectrometry based proteomics. Biol. Chem. 2017, 398, 687–699.
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965.
- Meyer, J.G. Fast Proteome Identification and Quantification from Data-Dependent Acquisition–Tandem Mass Spectrometry (DDA MS/MS) Using Free Software Tools. Methods Protoc. 2019, 2, 8.
- Prianichnikov, N.; Koch, H.; Koch, S.; Lubeck, M.; Heilig, R.; Brehmer, S.; Fischer, R.; Cox, J. MaxQuant Software for Ion Mobility Enhanced Shotgun Proteomics. Mol. Cell. Proteom. 2020, 19, 1058–1069.
- Rauniyar, N.; Yates, J.R. Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics. J. Proteome Res. 2014, 13, 5293–5309.
- Unwin, R.; Pierce, A.; Watson, R.B.; Sternberg, D.W.; Whetton, A.D. Quantitative Proteomic Analysis Using Isobaric Protein Tags Enables Rapid Comparison of Changes in Transcript and Protein Levels in Transformed Cells. Mol. Cell. Proteom. 2005, 4, 924–935.
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, A.T.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904.
- Li, J.; Cai, Z.; Bomgarden, R.D.; Pike, I.; Kuhn, K.; Rogers, J.C.; Roberts, T.M.; Gygi, S.P.; Paulo, J.A. TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing. J. Proteome Res. 2021, 20, 2964–2972.
- Leitner, A. A review of the role of chemical modification methods in contemporary mass spectrometry-based proteomics research. Anal. Chim. Acta 2018, 1000, 2–19.
- Hogrebe, A.; Von Stechow, L.; Bekker-Jensen, D.B.; Weinert, B.; Kelstrup, C.D.; Olsen, J.V. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9, 1045.
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116.
- Sonnett, M.; Yeung, E.; Wühr, M. Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster. Anal. Chem. 2018, 90, 5032–5039.
- Davies, D.R.; Gelinas, A.D.; Zhang, C.; Rohloff, J.C.; Carter, J.D.; O’Connell, D.; Waugh, S.M.; Wolk, S.K.; Mayfield, W.S.; Burgin, A.B.; et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. USA 2012, 109, 19971–19976.
- Ciampa, E.; Li, Y.; Dillon, S.; Lecarpentier, E.; Sorabella, L.; Libermann, T.A.; Karumanchi, S.A.; Hess, P.E. Cerebrospinal Fluid Protein Changes in Preeclampsia. Hypertension 2018, 72, 219–226.
- Larson, A.; Libermann, T.; Bowditch, H.; Das, G.; Diakos, N.; Huggins, G.; Rastegar, H.; Chen, F.; Rowin, E.; Maron, M.; et al. Plasma Proteomic Profiling in Hypertrophic Cardiomyopathy Patients before and after Surgical Myectomy Reveals Post-Procedural Reduction in Systemic Inflammation. Int. J. Mol. Sci. 2021, 22, 2474.
- Billing, A.M.; Ben Hamidane, H.; Bhagwat, A.M.; Cotton, R.J.; Dib, S.S.; Kumar, P.; Hayat, S.; Goswami, N.; Suhre, K.; Rafii, A.; et al. Complementarity of SOMAscan to LC-MS/MS and RNA-seq for quantitative profiling of human embryonic and mesenchymal stem cells. J. Proteom. 2017, 150, 86–97.
- Welton, J.L.; Brennan, P.; Gurney, M.; Webber, J.P.; Spary, L.; Gil Carton, D.; Falcón-Pérez, J.M.; Walton, S.P.; Mason, M.D.; Tabi, Z.; et al. Proteomics analysis of vesicles isolated from plasma and urine of prostate cancer patients using a multiplex, aptamer-based protein array. J. Extracell. Vesicles 2016, 5, 31209.
- Chirinos, J.A.; Cohen, J.B.; Zhao, L.; Hanff, T.; Sweitzer, N.; Fang, J.; Corrales-Medina, V.; Ammar, R.; Morley, M.; Zamani, P.; et al. Clinical and Proteomic Correlates of Plasma ACE2 (Angiotensin-Converting Enzyme 2) in Human Heart Failure. Hypertension 2020, 76, 1526–1536.
- Chan, M.Y.; Efthymios, M.; Tan, S.H.; Pickering, J.W.; Troughton, R.; Pemberton, C.; Ho, H.-H.; Prabath, J.-F.; Drum, C.L.; Ling, L.H.; et al. Prioritizing Candidates of Post–Myocardial Infarction Heart Failure Using Plasma Proteomics and Single-Cell Transcriptomics. Circulation 2020, 142, 1408–1421.
- Raafs, A.; Verdonschot, J.; Ferreira, J.P.; Wang, P.; Collier, T.; Henkens, M.; Björkman, J.; Boccanelli, A.; Clark, A.L.; Delles, C.; et al. Identification of sex-specific biomarkers predicting new-onset heart failure. ESC Hear. Fail. 2021, 8, 3512–3520.
- Wallentin, L.; Eriksson, N.; Olszowka, M.; Grammer, T.B.; Hagström, E.; Held, C.; Kleber, M.E.; Koenig, W.; März, W.; Stewart, R.A.H.; et al. Plasma proteins associated with cardiovascular death in patients with chronic coronary heart disease: A retrospective study. PLoS Med. 2021, 18, e1003513.
- Lau, E.S.; Paniagua, S.M.; Guseh, J.; Bhambhani, V.; Zanni, M.V.; Courchesne, P.; Lyass, A.; Larson, M.G.; Levy, D.; Ho, J.E. Sex Differences in Circulating Biomarkers of Cardiovascular Disease. J. Am. Coll. Cardiol. 2019, 74, 1543–1553.
- Huang, J.; Wu, N.; Xiang, Y.; Wu, L.; Li, C.; Yuan, Z.; Jia, X.; Zhang, Z.; Zhong, L.; Li, Y. Prognostic value of chemokines in patients with newly diagnosed atrial fibrillation. Int. J. Cardiol. 2020, 320, 83–89.
More