Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Amina Yu and Version 1 by Vinicius Cruzat.
Islet inflammation in T1DM is characterized by leukocyte infiltrates, in particular macrophages and T-cells which damage β-cells by release of cytokines, ROS and NO and also activation of death-receptor-mediated death pathways and subsequent phagocytosis. Production of cytokines such as INF-γ, TNFα and IL-1β act in synergy to promote elevation in concentration and increase in activity of NADPH oxidase and iNOS consequently increasing the formation of products including ROS and NO, respectively. The mechanism of action of INF-γ, TNFα and IL-1β involves stimulation of transcription factors including NFκB (in mouse islet β-cells).
- insulin
- metabolic reprogramming
- antidiabetic therapeutics
- glucose metabolism
- lipid metabolism
1. Introduction
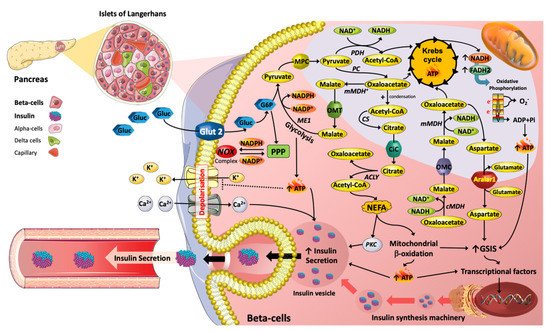
Figure 1. Metabolic reprograming mediated by nutrients with respect to insulin secretion machinery in Islet β cells. The metabolic pathways induced by glucose (Gluc) and lipids (e.g., NEFA) are key in the promotion of insulin secretion. For instance, TCA (tricarboxylic acid) cycle intermediates, such as Acetyl-CoA, Oxaloacetate, Malate and Citrate are essential in promoting insulin exocytosis. Moreover, TCA cycle intermediates can act as precursors for lipid signalling molecules that stimulate insulin vesicle trafficking and calcium influx, contributing to insulin secretion. Hence, changes in plasma glucose and NEFAs over physiological concentration ranges are essential for the regulation of β-cell function.
Pharmaceutical drug could induced metabolic adaptions/Reprogramming in β-Cells.
2. Antidiabetic Medications and Pancreatic β-Cell Metabolism
Recent studies indicate that metformin, GLP-1R agonists and the dipeptidyl peptidase-4 (DPP-4) inhibitors have direct effects on β-cells, promoting insulin secretion and/or limiting β-cell damage [101][1]. Metformin has been used therapeutically since the 1950s, is still the first-line treatment for T2DM as an effective antidiabetic agent, by reducing hepatic glucose production [101][1], restoring insulin secretion and protecting pancreatic β cells from lipotoxicity or glucotoxicity [102][2]. Metformin primarily acts on insulin-sensitive tissues. Observations from diabetic animal models and clinical trials have reported metformin can inhibit gluconeogenesis and suppress hepatic glucose production with improved insulin sensitivity in all peripheral tissues except the brain [103][3]. Metformin has pleiotropic actions in multiple organs or systematically [104][4] and can also partially ameliorate pancreatic β cell failure that occurs in T2DM [105][5].
Under homeostatic conditions, metformin does not promote or inhibit insulin secretion [106][6]; however, it reduces fasting plasma insulin concentration and enhances insulin sensitivity through reducing the activity of the pathway of hepatic gluconeogenesis, with a lesser effect on glycogenolysis [107][7]. Metformin directly modulates pancreatic β-cell function including moderate increase in insulin release, transcriptional regulation of β-cell signalling and cell viability, which is dependent on the presence of glucose [108][8], [109][9]. Metformin can restore insulin secretion previously suppressed by free fatty acids or high glucose in both human islets [110,111][10][11] and cell lines [105,112][5][12] due to improvements in glucose metabolism in the β-cell [113][13]. In experiments using Human islets cultured in high glucose concentrations, metformin reversed a reduction in GSIS associated with reduced ATP levels and a lower ATP/ADP ratio. Furthermore, metformin inhibited the activity of mitochondrial complex I and reversed ultrastructural alterations in β-cells exposed to high glucose levels [113,114][13][14]. Metformin prevented Ca2+-induced PTP opening in permeabilized and intact INS-1 cells to preserve β cell viability in hyperglycaemic conditions [115][15].
It has also been reported that metformin inhibits thapsigargin induced ER stress-induced apoptosis in a mouse pancreatic β cell line (NIT-1 cells) via upregulation of AMPK-PI3 kinase-JNK pathway [116][16]. Similarly, metformin protects rat insulinoma INS-1 cells from palmitate induced lipotoxic ER stress and apoptosis through decreasing JNK and p38MAPK phosphorylation via MAPK signalling pathways [117,118][17][18]. Moreover, metformin upregulates Aquaporin 7 in pancreatic islets (INS-1 cells) damaged by hyperglycaemic conditions through suppression of p38 and JNK mitogen activated protein kinase signalling to promote glycerol influx into β cells and subsequent promotion of insulin secretion [119][19].
Metformin prevented glucotoxicity through the reduction of oxidative and ER stress determined by a reduction in CD36 expression in pancreatic β-cells [120][20]. Pancreatic islets have relatively low expression of antioxidant enzymes which increases their risk of oxidative stress, see Section 2, Section 3 and Section 4 above [121][21]. Incubation of primary rat islets with metformin has been reported to restore insulin and pancreatic duodenal homeobox1 (Pdx) 1mRNA expression with recovery of GSIS and decreases in ROS production. Pancreatic duodenal homeobox -1 is a transcription factor that is expressed in β-cells and in duct cells of the pancreas stimulating cell proliferation and differentiation to form new islets and contributes to β-cell mass expansion and glucose metabolism induced via Akt signalling. [122,123][22][23]. Metformin also improved the antioxidant status (superoxide dismutase and catalase), glucose homeostasis and reduced inflammatory (TNF-α, IL-6) and ER stress (ATF4) markers in the pancreas of diabetic rats [124][24]. Furthermore, metformin increases gene expression in INS-1 β cells and mouse islet cells of GLP-1R, GIPR, and G protein-coupled receptor 40 (GPR40), and peroxisome-proliferator activated receptor α (PPARα) [125,126,127][25][26][27].
It is well established that failure of monotherapy can occur with metformin use in clinical practice [128][28]. Metformin was compared to rosiglitisone and glyburide (sulphonylurea) for control of hyperglycaemia, assessed over 5 years to determine the durability of each monotherapy [129][29]. β-cell function declined over 6-months in all three groups with the annual rate of decline highest in the glyburide group (a decrease of 6.1%), intermediate in the metformin group (a decrease of 3.1%), and least in the rosiglitazone group (a decrease of 2.0%). Metformin is currently prescribed when 3 months of lifestyle and dietary interventions have not reduced hyperglycaemia; however, as perturbations in β-cell function are already causing impaired glucose tolerance, it has been suggested that metformin therapy should be implemented earlier with lifestyle interventions to preserve β-cell function [130][30]. When monotherapy fails to control hyperglycaemia, agents with different pharmacological actions are combined to control blood glucose levels in T2DM patients and prevent chronic hyperglycaemia and the development of comorbid macro and microvascular diseases. In contrast, combinations of medications should be considered with respect to optimising β-cell metabolism and ultimately function, particularly during gluco- and lipotoxic conditions.
The second class of drugs that has historically been prescribed with metformin are the sulphonylureas, which bind to the ATP-sensitive potassium channels (KATP) to depolarise pancreatic β-cells and stimulate insulin release (see Marrano [101][1] for review of anti-diabetic drugs). Long-term exposure of the β-cells to sulphonylureas, both in in vitro and in vivo experiments, has been reported to decrease the extent of stimulated release of insulin due to desensitisation to this class of antidiabetic agent. For example, prolonged treatment of rodent BRIN-BD11 cells with glibenclamide reduced the acute insulinotropic actions of glucose [131][31]. Glibenclamide has also been demonstrated to increase apoptosis in human pancreatic islet cells [132][32]. The newer classes of antidiabetic agents include the thiazolidinediones which are peroxisome proliferator activated gamma receptor (PPARγ) activators that stimulate intracellular production of mediators that improve insulin sensitivity in tissues and the sodium-glucose transport protein 2 (SGLT2) inhibitors prevent renal absorption of glucose to increase excretion of glucose and reduce hyperglycaemia in patients with T2DM. Both of these classes of anti-diabetic agents can indirectly protect β-cell function through reducing hyperglycaemia. Dapagliflozin, a SGLT2 inhibitor, has been reported to decrease blood glucose concentrations and through upregulation of insulin and GLP-1 levels in T2DM mice, exhibit protective effects on the β-cells and enhanced β-cell replication [133][33].
Finally, GLP-1 receptors agonists (as previously discussed) and the DPP-4 inhibitors such as sitagliptin which inhibit breakdown of GLP-1 and GIP to elevate plasma levels of these hormones to stimulate insulin secretion and inhibit glucagon release, have the capacity to directly modify metabolic activity in β-cells. DPP-4 inhibition, through elevations in GLP-1, has been shown to activate CREB in insulin positive β-cells leading to increased Bcl-2, BIRC3 levels to inhibit apoptosis and elevate IRS-2, an adapter protein involved in insulin signalling [134][34].
3. Pancreatic β-Cell G-Protein Coupled Receptors and Cell Metabolism
Many β-cell G-protein coupled receptors need to be explored for their capacity to modulate insulin secretion [135][35]. Well established examples of islet GPCR are the receptors for the classical autonomic nervous system neurotransmitters such as acetylcholine (M3 muscarinic receptors) and noradrenaline (β2- and α2-adrenoceptors) which modulate insulin release under different conditions. Islet GPCRs that are sensitive to fatty acid ligands include the GPR40 and GPR119 and the receptors for the incretin hormones include GLP-1 and GIP as previously discussed. Islet GPCR for neuropeptides includes pituitary adenylate cyclase-activating polypeptide (PACAP), vasoactive intestinal polypeptide (VIP; PAC1 and VPAC2 receptors), cholecystokinin (CCKA receptors) and neuropeptide Y (NPY Y1 receptors). Other islet GPCR include cannabinoid receptors (CB1 receptors), vasopressin receptors (V1B receptors) and the purinergic receptors (P2Y receptors).
GPCRs in pancreatic β-cells are coupled to several signalling cascades including the cAMP/PKA/EPAC, and the inositol triphosphate (IP3)/diacylglycerol (DAG) pathways as well as changes to protein phosphorylation and protein acylation. In pancreatic β-cells, Gαs mediates increases in intracellular cAMP associated with increased insulin secretion, while Gαi mediates decreases in intracellular cAMP and inhibition of insulin secretion. Gαq mediates increases in IP3 and DAG production through the activation of phospholipase C (PLC) associated with increased release of Ca2+ from the ER and enhanced insulin secretion [136,137][36][37]. The M3 muscarinic receptors are functionally expressed in β-cells and enhance insulin secretion when blood glucose levels are elevated via coupling to Gαq activated PKC which raises intracellular Ca2+ levels. A positive allosteric modulator of M3 muscarinic receptors has been shown to restore glucose homeostasis in diabetic, glucose intolerant mice by promoting insulin secretion and may be a target for future clinical treatment of T2DM [138][38]. The long-chain fatty acid receptor GPR40 plays an important role in potentiation of GSIS from pancreatic β-cells. Activation of GPR40 by long-chain free fatty acids such as palmitate or oleic acid increases Ca2+ release from the ER by activating inositol 1,4,5-triphosphate (IP3) receptors [139][39]. Indeed, elevated Ca2+ levels can stimulate metabolism mainly though activation of mitochondrial TCA cycle activity and subsequent elevation in ATP levels
G-protein coupled receptor-55 (GPR55), a novel endocannabinoid receptor [140][40], is activated by phytochemicals, endocannabinoids and small synthetic cannabinoids [140,141,142][40][41][42]. In the pancreas, both human and rodent islets express GPR55 as indicated from gene and protein expression studies [142,143,144][42][43][44]. Furthermore, immunohistochemical analysis of rat and mouse pancreas sections demonstrated GPR55 expression specifically in insulin-secreting β-cells, but not in glucagon- or somatostatin-releasing α- and δ-cells, respectively [142[42][43],143], and have been identified as an anti-diabetic target [141,145][41][45]. The metabolic functionality of GPR55 ligands was investigated using CRISPR/Cas9 gene editing to determine their regulatory role in β cell and incretin-secreting enteroendocrine cell function. Atypical and endogenous endocannabinoid ligands stimulate insulin secretion in rodent (BRIN-BD11) and human (1.1B4) β cells through upregulation of insulin mRNA. As demonstrated in a clonal GPR55 knockout β cell line, GPR55 agonists enhance insulin secretion in β -cells, GIP and GLP-1 release from incretin-secreting enteroendocrine cells [146][46]. The GPR55 is specific for cannabinoid endogenous agonists (endocannabinoids) and non-cannabinoid fatty acids including L-α-lysophosphatidylinositol [147][47], and couples to Gα12/13 and Gαq proteins, leading to enhancing intracellular Ca2+, ERK1/2 phosphorylation and Rho kinase [140][40]. Primary studies have shown that activation of GPR55 receptors in pancreatic β-cells augments intracellular Ca2+ release, indicating a role for IP3 and intracellular calcium stores through PLCβ signalling [145,147,148,149,150,151][45][47][48][49][50][51]. Activation of the GPR55 also upregulates antiapoptotic genes such as Bcl-2 and Bcl-xL, reduces ER stress-mediated apoptosis and activation of the transcription factor cAMP response element binding to promote β-cell survival [146,148][46][48].
4. Contribution of Insulin, and Therapeutic Availability, to Metabolic Function of β-Cells
β-cells express insulin receptors, and autocrine insulin signalling is important for maintaining β-cell function by regulating cell proliferation, apoptosis and gene transcription [149][49]; however, the evidence is conflicting as to whether insulin mediates positive feedback of its own secretion in human islets [101][1]. Insulin increases the cytosolic Ca2+ concentration ([Ca2+]i) in rodent and human β-cells [150,151,152][50][51][52] by releasing Ca2+ from thapsigargin- and Nicotinic acid adenine dinucleotide phosphate (NAADP)-sensitive stores [151][51] but with minimal effects on membrane potential. Inhibition of insulin receptors reduces insulin secretion induced by other secretagogues, demonstrating that the β cell insulin receptor can provoke positive feedback for insulin secretion [150][50]. Increased [Ca2+]i was not accompanied by a stimulation of insulin release; therefore, IR-mediated signalling may affect other processes that regulate intracellular insulin release levels. In contrast, insulin (at concentrations comparable to those measured in plasma) has been found to inhibit glucose-induced Ca2+ oscillations in mouse islet cells and activate KATP channels to cause membrane hyperpolarization, suggesting that locally released insulin might generate a negative feedback signal within the islet during basal conditions. [153][53]. As a possible underlying mechanism, it was reported that insulin acutely activates PI3 kinase and increases PIP3 formation in an autocrine manner [154][54]. Acute disruption of insulin signalling using the tyrosine kinase inhibitor genistein in mouse-isolated islets [155][55] or the PI3 kinase inhibitor wortmannin in rat-isolated perfused islets [156][56] has also been reported to potentiate GSIS. As stated above, elevated Ca2+ levels can stimulate metabolism mainly though activation of mitochondrial TCA cycle (Ca2+ sensitive) enzyme activity and subsequent elevation in ATP levels.
References
- Marrano, N.; Biondi, G.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Functional loss of pancreatic islets in type 2 diabetes: How can we halt it? Metabolism 2020, 110, 154304.
- Yang, X.; Xu, Z.; Zhang, C.; Cai, Z.; Zhang, J. Metformin, beyond an insulin sensitizer, targeting heart and pancreatic β cells. Biochim. Biophys. Acta Mol. Bas. Dis. 2017, 1863, 1984–1990.
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270.
- El Messaoudi, S.; Rongen, G.A.; de Boer, R.A.; Riksen, N.P. The cardioprotective effects of metformin. Curr. Opin. Lipidol. 2011, 22, 445–453.
- Jiang, Y.; Huang, W.; Wang, J.; Xu, Z.; He, J.; Lin, X.; Zhou, Z.; Zhang, J. Metformin plays a dual role in MIN6 pancreatic β cell function through AMPK-dependent autophagy. Int. J. Biol. Sci. 2014, 10, 268–277.
- Leclerc, I.; Woltersdorf, W.W.; da Silva Xavier, G.; Rowe, R.L.; Cross, S.E.; Korbutt, G.S.; Rajotte, R.V.; Smith, R.; Rutter, G.A. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: Impact on glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1023–E1031.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investg. 2001, 108, 1167–1174.
- Hashemitabar, M.; Bahramzadeh, S.; Saremy, S.; Nejaddehbashi, F. Glucose plus metformin compared with glucose alone on β-cell function in mouse pancreatic islets. Biomed. Rep. 2015, 3, 721–725.
- Kitabchi, A.E.; Temprosa, M.; Knowler, W.C.; Kahn, S.E.; Fowler, S.E.; Haffner, S.M.; Andres, R.; Saudek, C.; Edelstein, S.L.; Arakaki, R.; et al. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the diabetes prevention program: Effects of lifestyle intervention and metformin. Diabetes 2005, 54, 2404–2414.
- Lupi, R.; Del Guerra, S.; Fierabracci, V.; Marselli, L.; Novelli, M.; Patanè, G.; Boggi, U.; Mosca, F.; Piro, S.; Del Prato, S.; et al. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes 2002, 51 (Suppl. S1), S134–S137.
- Lupi, R.; Del Guerra, S.; Tellini, C.; Giannarelli, R.; Coppelli, A.; Lorenzetti, M.; Carmellini, M.; Mosca, F.; Navalesi, R.; Marchetti, P. The biguanide compound metformin prevents desensitization of human pancreatic islets induced by high glucose. Eur. J. Pharmacol. 1999, 364, 205–209.
- Patanè, G.; Piro, S.; Rabuazzo, A.M.; Anello, M.; Vigneri, R.; Purrello, F. Metformin restores insulin secretion altered by chronic exposure to free fatty acids or high glucose: A direct metformin effect on pancreatic beta-cells. Diabetes 2000, 49, 735–740.
- Masini, M.; Anello, M.; Bugliani, M.; Marselli, L.; Filipponi, F.; Boggi, U.; Purrello, F.; Occhipinti, M.; Martino, L.; Marchetti, P.; et al. Prevention by metformin of alterations induced by chronic exposure to high glucose in human islet beta cells is associated with preserved ATP/ADP ratio. Diabetes Res. Clin. Pract. 2014, 104, 163–170.
- González-Barroso, M.M.; Anedda, A.; Gallardo-Vara, E.; Redondo-Horcajo, M.; Rodríguez-Sánchez, L.; Rial, E. Fatty acids revert the inhibition of respiration caused by the antidiabetic drug metformin to facilitate their mitochondrial β-oxidation. Biochim. Biophys. Acta 2012, 1817, 1768–1775.
- Lablanche, S.; Cottet-Rousselle, C.; Lamarche, F.; Benhamou, P.Y.; Halimi, S.; Leverve, X.; Fontaine, E. Protection of pancreatic INS-1 β-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Deat. Dis. 2011, 2, e134.
- Jung, T.W.; Lee, M.W.; Lee, Y.J.; Kim, S.M. Metformin prevents endoplasmic reticulum stress-induced apoptosis through AMPK-PI3K-c-Jun NH2 pathway. Biochem. Biophys. Res. Commun. 2012, 417, 147–152.
- Simon-Szabó, L.; Kokas, M.; Mandl, J.; Kéri, G.; Csala, M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS ONE 2014, 9, e97868.
- Dai, Y.L.; Huang, S.L.; Leng, Y. AICAR and Metformin Exert AMPK-dependent Effects on INS-1E Pancreatic β-cell Apoptosis via Differential Downstream Mechanisms. Int. J. Biol. Sci. 2015, 11, 1272–1280.
- He, X.; Gao, F.; Hou, J.; Li, T.; Tan, J.; Wang, C.; Liu, X.; Wang, M.; Liu, H.; Chen, Y.; et al. Metformin inhibits MAPK signaling and rescues pancreatic Aquaporin 7 expression to induce insulin secretion in type 2 diabetes mellitus. J. Biol. Chem. 2021, 297, 101002.
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30.
- Modak, M.A.; Parab, P.B.; Ghaskadbi, S.S. Control of hyperglycemia significantly improves oxidative stress profile of pancreatic islets. Islets 2011, 3, 234–240.
- McKinnon, C.M.; Docherty, K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia 2001, 44, 1203–1214.
- Jara, M.A.; Werneck-De-Castro, J.P.; Lubaczeuski, C.; Johnson, J.D.; Bernal-Mizrachi, E. Pancreatic and duodenal homeobox-1 (PDX1) contributes to β-cell mass expansion and proliferation induced by Akt/PKB pathway. Islets 2020, 12, 32–40.
- Obafemi, T.O.; Jaiyesimi, K.F.; Olomola, A.A.; Olasehinde, O.R.; Olaoye, O.A.; Adewumi, F.D.; Afolabi, B.A.; Adewale, O.B.; Akintayo, C.O.; Ojo, O.A. Combined effect of metformin and gallic acid on inflammation, antioxidant status, endoplasmic reticulum (ER) stress and glucose metabolism in fructose-fed streptozotocin-induced diabetic rats. Toxicol. Rep. 2021, 8, 1419–1427.
- Pan, Q.R.; Li, W.H.; Wang, H.; Sun, Q.; Xiao, X.H.; Brock, B.; Schmitz, O. Glucose, metformin, and AICAR regulate the expression of G protein-coupled receptor members in INS-1 beta cell. Horm. Metab. Res. 2009, 41, 799–804.
- Cho, Y.M.; Kieffer, T.J. New aspects of an old drug: Metformin as a glucagon-like peptide 1 (GLP-1) enhancer and sensitiser. Diabetologia 2011, 54, 219–222.
- Maida, A.; Lamont, B.J.; Cao, X.; Drucker, D.J. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-α in mice. Diabetologia 2011, 54, 339–349.
- Brown, J.B.; Conner, C.; Nichols, G.A. Secondary failure of metformin monotherapy in clinical practice. Diabetes Care 2010, 33, 501–506.
- Kahn, S.E.; Haffner, S.M.; Heise, M.A.; Herman, W.H.; Holman, R.R.; Jones, N.P.; Kravitz, B.G.; Lachin, J.M.; O’Neill, M.C.; Zinman, B.; et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443.
- Matthews, D.A.-O.; Del Prato, S.; Mohan, V.; Mathieu, C.; Vencio, S.; Chan, J.C.N.; Stumvoll, M.; Paldánius, P.M. Insights from VERIFY: Early Combination Therapy Provides Better Glycaemic Durability Than a Stepwise Approach in Newly Diagnosed Type 2 Diabetes. Diabetes Ther. 2020, 11, 2465–2476.
- Ball, A.J.; Flatt, P.R.; McClenaghan, N.H. Desensitization of sulphonylurea- and nutrient-induced insulin secretion following prolonged treatment with glibenclamide. Eur. J. Pharmacol. 2000, 408, 327–333.
- Maedler, K.; Carr, R.D.; Bosco, D.; Zuellig, R.A.; Berney, T.; Donath, M.Y. Sulfonylurea induced beta-cell apoptosis in cultured human islets. J. Clin. Endocrinol. Metab. 2005, 90, 501–506.
- Wei, R.; Cui, X.; Feng, J.; Gu, L.; Lang, S.; Wei, T.; Yang, J.; Liu, J.; Le, Y.; Wang, H.; et al. Dapagliflozin promotes beta cell regeneration by inducing pancreatic endocrine cell phenotype conversion in type 2 diabetic mice. Metabolism 2020, 111, 154324.
- Pugazhenthi, S.; Qin, L.; Bouchard, R. Dipeptidyl peptidase-4 inhibition in diabetic rats leads to activation of the transcription factor CREB in β-cells. Eur. J. Pharmacol. 2015, 755, 42–49.
- Delobel, M.; Dalle, S. G-protein–coupled receptors controlling pancreatic β-cell functional mass for the treatment of type 2 diabetes. Curr. Opin. Endocr. Metab. Res. 2021, 16, 113–118.
- Winzell, M.S.; Ahrén, B. G-protein-coupled receptors and islet function-implications for treatment of type 2 diabetes. Pharmacol. Ther. 2007, 116, 437–448.
- Persaud, S.J. Islet G-protein coupled receptors: Therapeutic potential for diabetes. Curr. Opin. Pharmacol. 2017, 37, 24–28.
- Zhu, L.; Rossi, M.; Cohen, A.; Pham, J.; Zheng, H.; Dattaroy, D.; Mukaibo, T.; Melvin, J.E.; Langel, J.L.; Hattar, S.; et al. Allosteric modulation of β-cell M(3) muscarinic acetylcholine receptors greatly improves glucose homeostasis in lean and obese mice. Proc. Natl. Acad. Sci. USA 2019, 116, 18684–18690.
- Usui, R.; Yabe, D.; Fauzi, M.; Goto, H.; Botagarova, A.; Tokumoto, S.; Tatsuoka, H.; Tahara, Y.; Kobayashi, S.; Manabe, T.; et al. GPR40 activation initiates store-operated Ca(2+) entry and potentiates insulin secretion via the IP3R1/STIM1/Orai1 pathway in pancreatic β-cells. Sci. Rep. 2019, 9, 15562.
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101.
- Tudurí, E.; Imbernon, M.; Hernández-Bautista, R.J.; Tojo, M.; Fernø, J.; Diéguez, C.; Nogueiras, R. GPR55: A new promising target for metabolism? J. Mol. Endocrinol. 2017, 58, R191–R202.
- McKillop, A.M.; Moran, B.M.; Abdel-Wahab, Y.H.; Flatt, P.R. Evaluation of the insulin releasing and antihyperglycaemic activities of GPR55 lipid agonists using clonal beta-cells, isolated pancreatic islets and mice. Br. J. Pharmacol. 2013, 170, 978–990.
- Romero-Zerbo, S.Y.; Rafacho, A.; Diaz-Arteaga, A.; Suarez, J.; Quesada, I.; Imbernon, M.; Ross, R.A.; Dieguez, C.; Fonseca, F.R.d.; Nogueiras, R. A role for the putative cannabinoid receptor GPR55 in the islets of Langerhans. J. Endocrinol. 2011, 211, 177–185.
- Liu, B.; Song, S.; Ruz-Maldonado, I.; Pingitore, A.; Huang, G.C.; Baker, D.; Jones, P.M.; Persaud, S.J. GPR55-dependent stimulation of insulin secretion from isolated mouse and human islets of Langerhans. Diabetes Obes. Metab. 2016, 18, 1263–1273.
- Moran, B.M.; Flatt, P.R.; McKillop, A.M. G protein-coupled receptors: Signalling and regulation by lipid agonists for improved glucose homoeostasis. Acta Diabetol. 2016, 53, 177–188.
- McCloskey, A.G.; Miskelly, M.G.; Moore, C.B.T.; Nesbit, M.A.; Christie, K.A.; Owolabi, A.I.; Flatt, P.R.; McKillop, A.M. CRISPR/Cas9 gene editing demonstrates metabolic importance of GPR55 in the modulation of GIP release and pancreatic beta cell function. Peptides 2020, 125, 170251.
- Simcocks, A.C.; O’Keefe, L.; Jenkin, K.A.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. A potential role for GPR55 in the regulation of energy homeostasis. Drug Discov. Today 2014, 19, 1145–1151.
- Vong, C.T.; Tseng, H.H.L.; Kwan, Y.W.; Lee, S.M.; Hoi, M.P.M. G-protein coupled receptor 55 agonists increase insulin secretion through inositol trisphosphate-mediated calcium release in pancreatic β-cells. Eur. J. Pharmacol. 2019, 854, 372–379.
- Leibiger, I.B.; Leibiger, B.; Berggren, P.O. Insulin signaling in the pancreatic beta-cell. Annu. Rev. Nutr. 2008, 28, 233–251.
- Aspinwall, C.A.; Lakey, J.R.; Kennedy, R.T. Insulin-stimulated insulin secretion in single pancreatic beta cells. J. Biol. Chem. 1999, 274, 6360–6365.
- Johnson, J.D.; Misler, S. Nicotinic acid-adenine dinucleotide phosphate-sensitive calcium stores initiate insulin signaling in human beta cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14566–14571.
- Luciani, D.S.; Johnson, J.D. Acute effects of insulin on beta-cells from transplantable human islets. Mol. Cell Endocrinol. 2005, 241, 88–98.
- Khan, F.A.; Goforth, P.B.; Zhang, M.; Satin, L.S. Insulin activates ATP-sensitive K(+) channels in pancreatic beta-cells through a phosphatidylinositol 3-kinase-dependent pathway. Diabetes 2001, 50, 2192–2198.
- Hagren, O.I.; Tengholm, A. Glucose and insulin synergistically activate phosphatidylinositol 3-kinase to trigger oscillations of phosphatidylinositol 3,4,5-trisphosphate in beta-cells. J. Biol. Chem. 2006, 281, 39121–39127.
- Jonas, J.C.; Plant, T.D.; Gilon, P.; Detimary, P.; Nenquin, M.; Henquin, J.C. Multiple effects and stimulation of insulin secretion by the tyrosine kinase inhibitor genistein in normal mouse islets. Br. J. Pharmacol. 1995, 114, 872–880.
- Zawalich, W.S.; Zawalich, K.C. Effects of glucose, exogenous insulin, and carbachol on C-peptide and insulin secretion from isolated perifused rat islets. J. Biol. Chem. 2002, 277, 26233–26237.
More