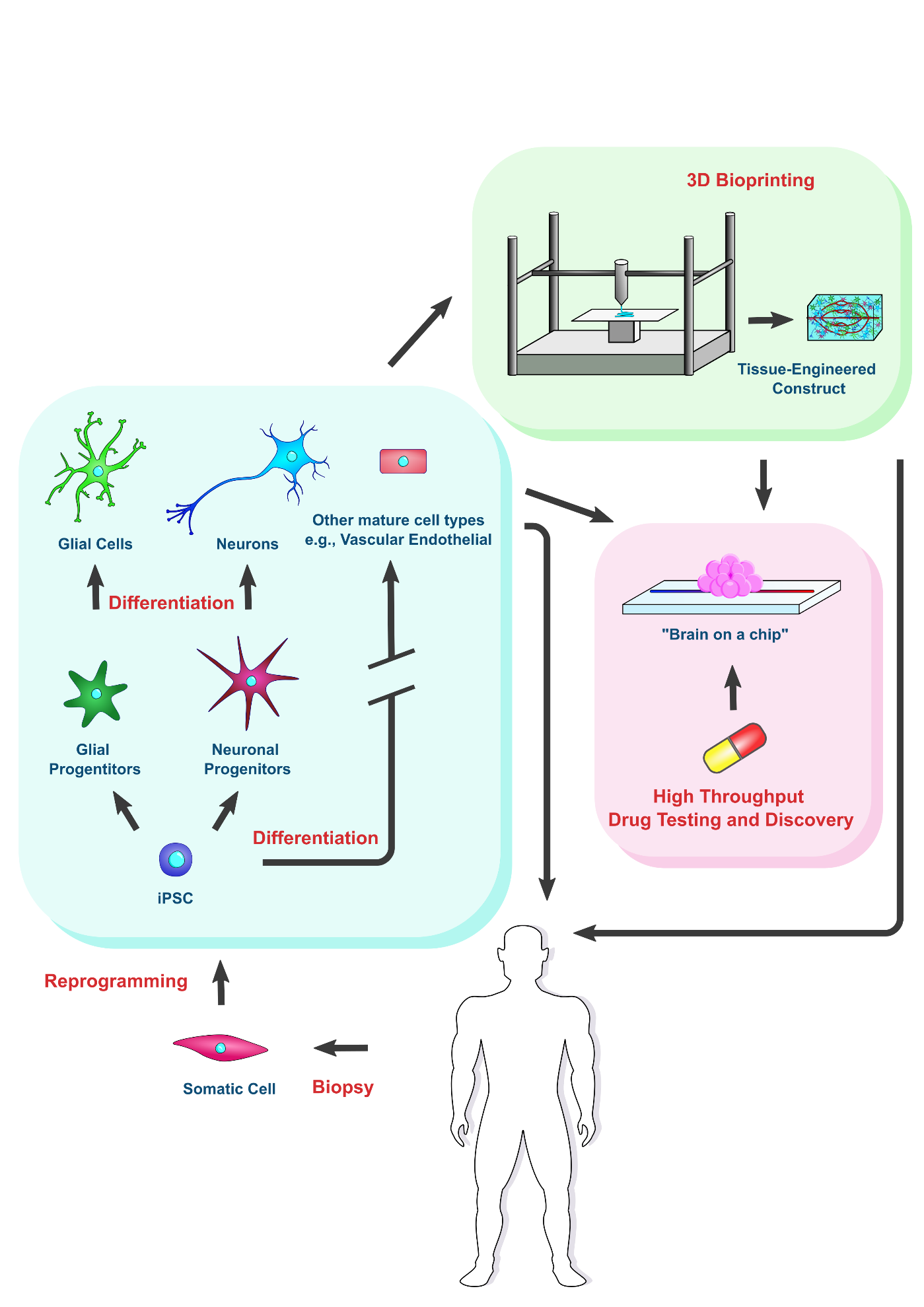
Human pluripotent stem cells (hPSC
onventionally, Human pluripotent stem cells (hPSCs)s) are receiving much attention in the field of tissue regeneration. They can be differentiated into any cell type in the human body and subsequently developed into heterogeneous tissues or organs in vitro for implantation. Conventionally, hPSCs are harvested from surplus embryos that were generated after in vitro fertilization, known as embryonic stem cells (ESCs). A number of clinical trials employing human embryonic stem cell (hESC)-derived tissue have been performed. Many of them are focused on the treatment of macular degeneration, and have shown promise in improving visual acuity
[1][2][3][4][17,18,19,20]. Likewise, a number of studies using ESCs were carried out in an attempt to treat ischemia
[5][21] and even pulmonary fibrosis caused by COVID-19
[6][22]. Implantation of mesencephalic fetal tissue into the putamen has also exhibited promising results in the treatment of neurodegenerative diseases, particularly in younger patients
[7][23]. Clinical trials have recently been completed for the treatment of Alzheimer’s using mesenchymal stem cell therapy
[8][24]. The majority of these trials, however, required parallel immunosuppressive treatment, and obtaining these cells required harvesting them from a blastocyst, which presents an ethical issue: an embryo with a unique genome has to be destroyed, which would have otherwise developed into a human being
[9][25]. If then used for regenerative implantation, fetal stem cells carry a high risk of invoking an immune rejection and graft-versus-host disease (GVHD) upon implantation, which is why immunosuppressive drugs would have to be taken postimplantation
[10][26].
Other methods of obtaining pluripotent stem cells exist, such as somatic cell nuclear transfer (SCNT)
[11][27] and induction of parthenogenesis
[12][28]. The latter is a technique in which an unfertilized egg cell, or oocyte, is stimulated to divide. This can be undertaken at various points of meiosis. For example, a primary oocyte that still has a diploid nucleus may be stimulated to undergo mitosis. However, inherent problems with parthenogenesis, such as decreased protection against tumorigenicity owing to their homozygosity, as well as a lack of balanced epigenetic imprinting, are currently hurdles in its use for regenerative medicine
[13][29]. SCNT, on the other hand, involves transferring a diploid nucleus from a differentiated somatic cell into an enucleated oocyte. The inefficiency and low availability of oocytes, however, hinder the prospect of using SCNT for regenerative medicine
[14][30].
The groundbreaking discovery of iPSCs in 2006 by the Yamanaka group opened the door to a sustainable source of pluripotent stem cells, bypassing the ethical concerns and many of the technical hurdles associated with using conventional methods
[15][31]. Using iPSCs offers an abundant source of stem cells that are autologous to a patient, removing the need for an immunosuppressive regimen postimplant, and allowing more accurate simulation of native tissue in vitro. Cells generated using this contemporary method would not be subject to immune rejection, as they are derived from the patient receiving the transplantation. This method revolves around the reprogramming of somatic cells, which, of course, has its own challenges, but nonetheless, remains a pivotal discovery for tissue engineering and shows promise in the treatment of neurodegenerative disease. Remarkably, plenty of studies have demonstrated that iPSCs and ESCs are molecularly and functionally equivalent
[16][17][32,33]. Importantly, iPSCs and ESCs exhibited similar lineage specification capability when they were compared side by side for the generation of a functional tissue or organ, including but not limited to neurons, pancreatic islet organoids, cardiomyocytes, and kidney micro-organoids
[18][19][20][21][22][23][24][25][34,35,36,37,38,39,40,41]. This pivotal feature makes iPSCs valuable sources for biomedical applications. However, variations in differentiation exist among the different iPSC lines, due in part to the reprogramming methods applied during the creation of iPSC lines and the individual genetic features of donor cells
[26][27][42,43]. Hence, iPSC line selection should be taken into account accordingly to develop strategies for cell therapy and specific disorder modeling
[28][44].
2. iPSCs for Regenerative Cell Therapies Applied to Alzheimer’s and Parkinson’s Treatment
Implantation can range from raw iPSC populations to homogenous progenitor cell populations, to complex tissue-engineered constructs consisting of multiple cell species, for recovering regions damaged by ND. A number of studies demonstrated successful implantation of raw iPSCs into Alzheimer’s animal disease models.
ItOne was ssuch study showed that, after injecting iPSCs into 5XFAD Alzheimer’s mice, the iPSCs differentiated into glial cells upon implantation. Particularly, microglia, oligodendrocytes, and astrocytes were generated from the injected cells in vivo. The amount of Aβ plaque deposition decreased, along with a decrease in the activity of beta and gamma secretases, and increases in oligodendrocyte-related gene expression in the iPSCs-treated mice (
Table 1)
[29][45]. Additionally, iPSC-treated mice performed better on cognitive tasks, as evaluated by effectiveness in completing a maze. It is worth noting that the iPSCs used
in this study were derived using a novel method, rendering the stem cells safer for transplantation and getting them closer to clinical trials. Typically, inducing somatic cells to reprogram into undifferentiated pluripotent stem cells required the introduction of exogenous genetic material via a viral vector
[15][31]. This would cause the overexpression of a particular set of transcription factors, coined Yamanaka factors, leading to dedifferentiation. However, introduction of these genes augments the tumorigenic potential of the obtained stem cells and their derived tissues.
Hence, in a previous study, Cho and his coworkers showed a protein-based method for inducing pluripotency without genetic modification, increasing safety, as compared to previous methods. It was performed via a streptolysin-mediated permeabilization protocol of proteins extracted from embryonic stem cells, which did not require long-term exposure of target cells to exogenous materials
[30][46].
Table 1. Examples of preclinical trials using iPSC-derived cells for AD and PD disease treatment.
Source of
iPSCs |
Neurodegenerative
Disease Treated |
Model |
Type of Cells |
Number |
Route of
Delivery |
Outcome |
Reference |
| Autologous, mouse skin fibroblasts |
AD |
in vivo: 5XFAD mice |
iPSCs |
100,000 |
Injection into
subiculum |
Decrease in Aβ plaque deposition and beta/gamma-secretase
activity |
[29] |
| Autologous, skin fibroblasts |
PD |
in vivo: Parkinsonian cynomolgus monkeys |
Dopaminergic neurons |
10–40 million |
Injection into four sites of
post-
commissural putamen |
Improvements in motor function and reinnervation by implanted neurons |
[31] |
| Autologous and allogeneic, skin fibroblasts |
PD |
in vivo: Parkinsonian rhesus monkeys |
Dopaminergic neurons |
5.5–22 million |
Injection into
basal ganglia |
Improvements in motor function consistent with reinnervation by
implanted neurons seen in autologous transplant group |
[32] |
| Human dermal fibroblast lines |
PD |
in vivo: immunodeficient 6-OHDA Parkinsonian mice |
Dopaminergic neuron progenitors |
100,000–300,000 |
Injection into
striatum |
Recovery of rotation
behavior, improvements on corridor, cylinder, stepping tests |
[33] |
A number of studies have shown the survivability of autologous iPSC-derived dopaminergic neurons or progenitors in vivo, which was achieved by transplantation into primate or murine PD models
[32][31][33][47,48,49]. Implantation of iPSC-derived dopaminergic neurons, which have been differentiated in culture, into the putamen of Parkinsonian cygnus monkeys resulted in reinnervation via the implanted neurons, and improvements in motor function
[31][48]. In another study, similarly, five Parkinsonian rhesus monkeys received autologous iPSC-derived dopaminergic neuron implants, and motor improvements were compared with those of another five Parkinsonian rhesus monkeys that received allogeneic transplantation. Midbrain dopaminergic neural progenitor cells were implanted into the four basal ganglia 1–3 years after induction of Parkinson’s via MPTP–HCL. Monkeys receiving autologous transplantation showed improvements in motor function as well as alleviation of mood disorder symptoms 24 months after surgery
[32][47]. However, a unilateral model was used, so improvements may have been augmented by the healthy side
[32][47], and no immunosuppression was used for the group receiving an allogeneic transplant. As improvements were correlated with the number of surviving dopaminergic neurons, suppression of the immune response to foreign dopaminergic neurons may have otherwise decreased their elimination, and allowed further alleviation of symptoms in monkeys receiving allogeneic transplants
[32][47]. Another study by the Song research group generated clinical-grade midbrain dopaminergic neural progenitors from an iPSC cell line. Implantation of 100,000–300,000 of these cells into the striatum of immunodeficient Parkinson-induced mice yielded significant recovery in motor function after 14 weeks, which was sustained after at least 52 weeks. To bring these cells closer to clinical grade, they were produced under good manufacturing practice (GMP) using a specific protocol with the following key points: an episomal vector, along with selected miRNA, improved reprogramming efficiency of fibroblasts to iPSCs. A “spotting” technique was implemented, where cultures were split into smaller “spots”, yielding higher cell viability during growth and differentiation. Preimplantation, whole genome sequencing, karyotyping, and quantitative real-time polymerase chain reaction (qRT-PCR) were performed to ensure that selected iPSCs did not carry any known tumorigenic mutations nor integration of the episomal plasmid. The progenitors were also treated with quercetin to eliminate any leftover undifferentiated stem cells to avoid the risk of tumorigenesis upon implantation
[33][49].
The Schweitzer research group at Massachusetts General Hospital implanted human autologous iPSC-derived neural progenitor cells into a human subject.
AIn preparation, a study was conducted to test the immunogenicity of iPSC-derived dopamine neural progenitor cells. They implanted midbrain dopaminergic progenitor cells generated from patient-derived iPSCs, as well as those from a different human iPSC line, into immune-deficient mice, K1 humanized mice, and mice with immune cells taken from the same patient (K1 mice had human immune cells but not from the patient). Both implant types survived in the immune-deficient mice, but were destroyed by the immune responses in K1 humanized mice, and only the patient-derived dopaminergic cells survived in the mice with the patient’s immune cells. The patient then received a graft of these dopaminergic progenitor cells to the putamen of both hemispheres at around 4 million cells per hemisphere. Throughout a period of 24 months following the initial operation, the patient showed increases of dopamine uptake by cells surrounding the implant site as evaluated by Flourodopa F18 (F-DOPA) positron emission tomography (PET). This was accompanied by improvements in Parkinsonian symptoms as evaluated by the Movement Disorder Society’s prescribed Unified Parkinson’s Disease Rating Scale (MDS–UPDRS), the Parkinson’s disease Questionnaire (PDQ-39), and the patient-recorded duration of “off times”
[34][50]. Despite the very small sample size of one person, unknown long-term effects, and change in medication dosage being a possible interfering factor,
it
washe study showed barely significant but promising results
[34][50].
IWhile previous st
waudies showed alleviated Parkinsonism in animal disease models upon implantation of iPSC-derived dopaminergic neuron progenitors
[32][33][47,49], this was the first
inst
roduceudy that implanted the dopaminergic progenitor cells into a human subject.
So far, iPSC-derived products for PD and AD have not undergone many clinical trials.
T Apart from this study, the only example of iPSC-derived implantation into humans involves an ongoing clinical trial in Japan, for the treatment of PD, which was documented
in a review by Ford and colleagues
[35][51]. In Australia and China, hESC-derived cells are also undergoing clinical trials for treatment of PD
[35][51]. Due to the aforementioned similarity between iPSCs and ESCs, it can be expected that iPSC-derived cells will also make their debut in clinical trials in the near future.
3. Using iPSC-Derived Organoids to Model Pathophysiology of Neurodegenerative Diseases
In addition to implantation for cell therapy, iPSCs can be used for the production of in vitro models of the afflicted region of the brain in ND patients. Since the donor’s iPSCs carry the same genotype as the donor, this is especially useful for understanding how genetic variations affect pathology, and can prove especially beneficial for personalized drug discovery aimed towards specific genetic variants. The pathophysiology of Alzheimer’s and Parkinson’s is affected by genetic variations from person to person, which plays a role in drug responsiveness. For example, it is known that the effectiveness of L-DOPA in treating Parkinson’s varies depending on genotype
[36][37][52,53], and it was even found that donepezil, a widely prescribed drug for AD, worsens Alzheimer’s symptoms in patients with a certain genotype
[38][54]. Hence, in vitro models encompassing the pharmacogenetic polymorphisms across the population may prove useful in developing personalized medication regimens. Disease models can greatly help in the quest to better understand the pathological progression of Parkinson’s and Alzheimer’s and to test drug candidates for safety and efficacy. For this purpose, neurodegenerative diseases can be induced in animals. However, animal models often do not recapitulate human in vivo pathophysiology accurately. Indeed, some drugs that have been deemed safe and effective in animal trials were found to show very different results in humans. Hence, tissues or organoids derived from human iPSCs, such as a “brain on a chip”, show great promise in closely recapitulating the human in vivo pathophysiology and in aiding in drug discovery.
As previously mentioned, it is crucial to observe not only neurons but also glial cells, and the interplay of both cell species, in the progression of ND. Yet another key component involves vascular cells, specifically at the blood–brain barrier (BBB). In vascular neurodegenerative disease, cytotoxicity occurs due to the breakdown of neurovascular units, which consist of a combination of vascular endothelial cells, pericytes, neurons, and neuroglia making up the BBB
[39][55]. A study by Jagadeesan and his coworkers exemplified the modeling of the BBB on a “brain-on-a-chip” device. This was performed by seeding two adjacent channels, separated by a semipermeable membrane, with iPSC-derived cells. One channel representing the neural side was seeded using “EZ spheres”, which are clusters of neural progenitor cells. Putting them through a differentiation protocol yielded a coculture of mature neurons, neural progenitors, and astrocytes (a subpopulation of neuroglia). The second channel was seeded with brain microvascular endothelial cells (BMECs), which adhered to extracellular matrix (ECM) proteins that were chemically attached to the polydimethylsiloxane (PDMS) semipermeable membrane between the two channels. The proteins were optimally selected for these specific cell types, a requirement for proper cell adhesion and functional development. The final cell populations were assessed via immunocytochemical analysis, and the permeability of the interface between the two channels was determined. Vascular endothelial cells formed tight junctions with each other, as is seen in the natural BBB, but this only occurred in the presence of the neural cell populations, highlighting the importance of cell–cell signaling for proper BBB formation. The final construct allowed perfusion of the “blood” channel that is lined with the microvascular endothelial cells for the purpose of testing the permeability of various drugs across the BBB
[40][56]. Since patient-specific iPSCs are used, the exact response of a patient’s neurovascular system on a drug can be ascertained. Thenature of this “organ-on-a-chip” technology, used in conjunction with patient-specific iPSCs, leaves the system open to improvements towards iPSC differentiation and inclusion of additional cellular subpopulations, such as pericytes
[40][56]. Prospectively, cells taken from an AD or PD patient can be used to accurately model in vivo pathogenesis.
The SNCA gene encodes the alpha-synuclein protein. It is strongly correlated to the onset of familial PD. Studies have demonstrated that a triplication of the SNCA gene causes an increase in the buildup of alpha-synuclein deposits in iPSC-derived neural progenitor cells, which inhibits their maturation
[41][57]. To build on this, Mohamed and his coworkers pioneered the use of 3D human midbrain organoids (hMOs) derived from a patient’s iPSCs for modeling SNCA triplication in order to investigate the effect of SNCA triplication on PD pathology. The organoids with triplication exhibited the expected Parkinson’s pathophysiology, with a lower number of dopaminergic neurons, and a decrease in size after 100 days, compared to an isogenic control. This method paves the way for more accurate disease modeling, as 3D midbrain organoids can survive for longer, allowing the deduction of age-dependent changes. Furthermore, the 3D organoids contain many types of cells, allowing closer mimicry of the cell–cell interactions in vivo
[42][58]. The Raja research group developed scaffold-free, 3D self-assembling neural organoids that demonstrate the hallmark signs of Alzheimer’s: tauopathy and Aβ accumulation. They also demonstrated “in vitro aging” of the organoids and their platform was highly responsive to drugs, allowing for observation of phenotypic changes relating to the application of beta or gamma secretase inhibitors
[43][5]. Using this 3D model, which more closely resembles actual Parkinsonian progression, would improve the discovery and testing of drugs moving towards preclinical drug validation. Nevertheless, it was noted that the exact correlation between the “in vitro aging” of the organoids and the maturation of an actual human brain is yet to be determined
[43][5], which is critical for further research.
A further study by the Mohamed research group developed a method aimed at addressing issues with the scale up of organoid production. They proposed the packaging of iPSC-derived brain organoids on 948-well, microfabricated disks of Matrigel. This method ameliorated the efficiency of hMO production on a large scale, yielding lower cost and batch-to-batch variability. It does not require automation, making it more accessible to labs with only basic equipment
[44][59].
With the production of neural constructs, the major issue of vascularization remains. A construct with cells further than 200–400 microns from surface contact with the culture medium must have an adequate vascular network to supply nutrients, remove waste, and transport cell signaling molecules, due to the diffusion limits. In a recent study, researchers coated whole-brain organoids derived from a patient’s iPSCs in a layer of Matrigel seeded with endothelial cells (ECs), which were derived from the same patient’s iPSCs. This resulted in angiogenesis of the organoid from the outside in. Implantation of the construct into a mouse model resulted in deeper penetration of the vascular network by the human ECs when compared to just in vitro vascularization. This is promising for the future of larger neural implants and organoids, which must overcome the lack of blood vessels in vitro
[45][60]. Nevertheless, more work is needed to determine the exact cell signal cascade that augmented this vascularization process in vivo. Its translation would allow growth of larger organoids in vitro using a specifically defined medium conducive to angiogenesis.