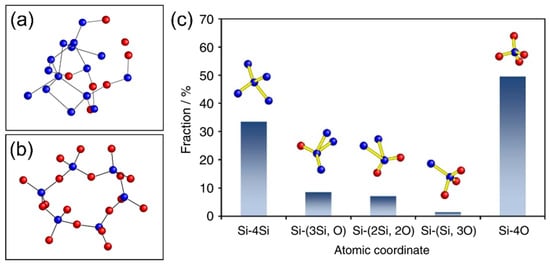
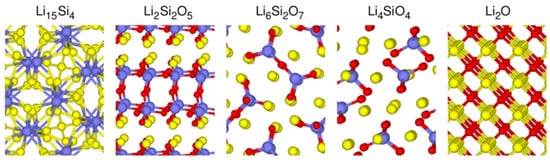
Since various lithium silicates have different crystalline structures and Li/Si ratios, their material properties and electrochemical properties are not the same. For instance, the Li diffusivities of compounds consisting of lithium, silicon, and oxide were calculated via the climbing-image nudged elastic band (CI-NEB) method, and the diffusivities ranked from low to high are: Li
6Si
2O
7 (1.8 × 10
−12) < Li
2Si
2O
5 (1.3 × 10
−11) < Li
4SiO
4 (1.1 × 10
−10) < Li
2O (1.8 × 10
−8) < Li
15Si
4 (8.8 × 10
−6)
[13]. According to the simulation results, fully-lithiated silicon has the highest diffusivity compared to all lithium silicates, and Li
2O has a Li diffusivity that is two orders of magnitude higher than that of other inactive lithium silicates
[13]. Another report demonstrated that at low Li concentrations, the diffusivity of Li
+ ions in lithium silicates is two orders of magnitude lower than that of lithium silicides (Li
xSi), but it rapidly increases with increasing Li content
[14]. Lithium silicates with high Li-ion diffusivity facilitate fast charge/discharge capability, high utilization of active materials, and low polarization/overpotential. In the subsections below,
we introduce tthe electrochemical properties and formation conditions of lithium silicates in anode materials
should be introduced. Simplified formation mechanisms of lithium silicates from SiO and SiO
2 can be shown as follows (assuming Li
xSi
yO
z species are generated before Li
xSi)
[15,16][15][16]:
3.1. Li4SiO4
As one of the major phases in lithiated a-SiO, Li
4SiO
4 is the most studied buffer material that minimizes the volume expansion of SiO anode
[4,5,11,17][4][5][11][17]. Li
4SiO
4 functions as an inactive material but can maintain structural integrity due to its strong mechanical strength. The capability of conducting Li
+ ions is very important for Li
4SiO
4 to serve as both an ionic conductor and a buffer material, which guarantees smooth electrochemical reactions. It was reported that, in terms of Li ionic conductivity, the conductivity of crystalline Li
4SiO
4 in SEI detected by atomic visualization is estimated to be 1000 times greater than that of Li
2O, according to molecular dynamic simulation
[18]. Pristine Li
4SiO
4 has been identified as either chemically or electrochemically inactive with lithium
[1]. This means that Li
4SiO
4 can be neither lithiated nor delithiated once formed
[1,19,20][1][19][20]. However, Hirose et al. demonstrated that Li
4SiO
4 can release Li
+ ions at a high potential (2.5 V vs. Li/Li
+)
[17,21][17][21]. Additionally, it is believed that Li
4SiO
4 is the lithium silicate with the highest Li/Si ratio that can be formed in an electrochemical cell. Coyle et al. adopted radio-frequency magnetron sputtering of both SiO
2 and Li
2O targets to make thin lithium silicate films with various compositions, and a lithium-rich Li
8SiO
6 (Li/Si = 8) phase was confirmed by inductively coupled optical emission spectrometry (ICP-OES), Fourier transform infrared (FTIR) spectroscopy, time-of-flight secondary-ion mass spectrometry (ToF-SIMS), and X-ray photoelectron spectroscopy (XPS)
[22]. However, there have been no electrochemical performance data reported on Li-rich lithium silicates with a Li/Si ratio higher than four.
Yamamura et al. found that neither crystalline Li
4SiO
4 nor SiO
2 can react with lithium metal, but amorphous SiO
2 showed Li-ion activity
[1]. It is believed that the SiO
4 tetrahedra in Li
4SiO
4 and SiO
2 are difficult to reduce by lithium metal and ions, but the dangling bonds and distorted SiO
4 tetrahedra in a-SiO
2 can facilitate reactions with lithium. The formation energy (E
f) of lithium inserting into the SiO
4 tetrahedra of crystalline SiO
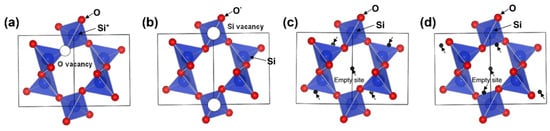
2 can be calculated. When lithium stays in the O vacancy (
Figure 3a), the E
f is 0.44 eV, requiring extra energy to be stabilized. When lithium is in the Si vacancy (
Figure 3b), the E
f is −3.66 eV, which represents a spontaneous exothermic reaction of Li
+ with O
−. This indicates that lithium has a high tendency to bond with oxygen due to the electrostatic attraction force originating from their opposite charges. In addition, when lithium is located at the empty sites in the structure shown in
Figure 3c,d, the E
f is 1.15 eV and 1.18 eV, respectively, implying the difficulty of creating bonds. Sivonxay et al. employed DFT and ab initio molecular dynamics (AIMD) methods to demonstrate that amorphous SiO
2 is thermodynamically driven to be lithiated, which shows that Li–O local environments are increasingly favored, as compared to Si–O bonding
[14]. Moreover, amorphous SiO
2 and SiO have disordered structures, including many vacancies, dangling bonds, and non-equivalent bond lengths, offering more reaction sites for lithiation. This explains why amorphous silicon oxides exhibit lithium activity, while crystalline Li
4SiO
4 and SiO
2 are considered chemically and electrochemically inert with lithium.
Figure 3. The crystalline structure of SiO
2 (
P3121 space group) drawn via the VESTA method (
a) with O vacancy, (
b) with Si vacancy, and (
c,
d) with empty sites. Reproduced with permission from ref.
[1]. Copyright 2011, The Ceramic Society of Japan.
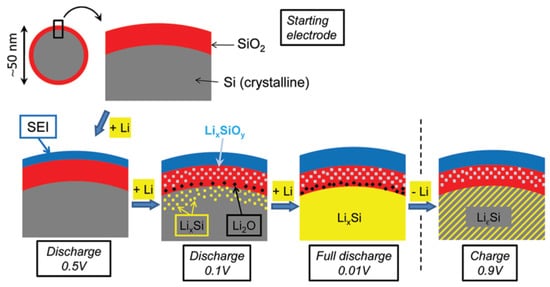
Lithium silicates were also discovered in the oxidized surface of silicon nanoparticles during cycling in LIBs
[23]. When pure silicon is in contact with air, a thin nanocoating of silica is formed on the silicon surface, which prevents further oxidation.
Figure 4 exhibits how the microstructure and composition of nano-sized Si with naturally formed SiO
2 change, along with discharge and charge conditions. When the silica-covered silicon anode is discharged to ~0.5 V, a solid electrolyte interface (SEI) layer is formed under the electrolyte reduction reactions. This layer passivates the anode surface to avoid further Li-ion and electrolyte consumption. While the Li/Si cell is deeply discharged, both Li
xSi
yO
x and Li
2O are formed in the surface oxide region, and Li
xSi is formed in the core silicon region. By analyzing the signal of Si 2p spectra, it was confirmed that Li
4SiO
4 is the main lithium silicate phase
[23]. After the cell is recharged to 0.9 V, Li
2O is gone, but Li
4SiO
4 still exists in the structure, evidencing the reversibility of Li
2O and the irreversibility of Li
4SiO
4. On the surface of a silicon anode, Li
4SiO
4 remains in the protective layer, remaining unchanged during cycling, similar to a-SiO and disproportionated SiO
x anodes
[12,13][12][13].
Figure 4. Microstructure of nano-sized Si with naturally formed SiO
2 on the surface. Composition changes during shallow and deep discharge and full recharge. Reproduced with permission from ref.
[23]. Copyright 2012, ACS Publications.
The formation of Li
4SiO
4 in SiO/SiO
X-based anodes occurs during the first discharge process at <0.24 V vs. Li/Li
+, and the crystalline phase remains in the anode structure regardless of cell recharge or cycling, showing strong irreversibility
[19,20,24][19][20][24]. Besides the electrochemically generated Li
4SiO
4 in situ, Li
4SiO
4 can be synthesized by the thermal reaction of SiO and lithium hydroxide cofired at 550 °C
[25,26][25][26]. A simple hydrothermal synthesis method (175 °C) using fumed silica and lithium hydroxide can also produce Li
4SiO
4 [27]. A citric-acid-based sol-gel route (also with fumed silica and LiOH) was developed to synthesize macroporous Li
4SiO
4 with a carbon coating
[28]. Due to the stability of electrochemical cells and the synthesizability of Li
4SiO
4, it can serve as a promising protective buffer material for silicon and lithium metal anodes, which will be discussed in the next section.
3.2. Li2SiO3
Li
2SiO
3 has lithium activity, unlike Li
4SiO
4, with a reversible capacity of <200 mA h g
−1 [29]. Yang et al. utilized graphene to prepare a composite material with Li
2SiO
3 as the anode, showing good cycle life
[29]. Pristine Li
2SiO
3 has sloping charge/discharge curves with an average voltage above 1 V, and it can be reversibly cycled in a half cell with lithium metal. Li
2SiO
3 can be formed by heating a graphite/Li
4SiO
4 mixture at 600 °C
[30]. A graphite composite sample with Li
2SiO
3 obtained a higher capacity than that with Li
4SiO
4, although larger first-cycle capacity loss was also found in an anode sample with Li
2SiO
3. Xiao et al. pointed out that Li
2SiO
3 can improve Li-ion diffusion behavior for better rate performance and that co-existing Li
2CO
3 favors the growth of a denser and more conductive SEI layer
[30]. Li
2SiO
3 has also been synthesized in a graphene composite material with Li
4Ti
5O
12 or Li
2SnO
3 [31,32][31][32]. Both anode composite materials with Li
2SiO
3 have better capacity and rate performance due to their lithium activity and good ionic conductivity.
Li
2SiO
3 was also found in lithiated SiO/SiO
x-based anode materials and electrodes under both shallow and deep lithiation conditions
[1,33,34][1][33][34]. Both Li
4SiO
4 and Li
2SiO
3 can be generated either by chemical lithiation with lithium foil or powder at 600 °C–800 °C (
Figure 5a) or electrochemical lithiation within a half cell with a lithium metal. Since Li
4SiO
4 is an inert material for lithiation/delithiation and Li
2SiO
3 is active to Li
+ ions with good reversibility, there should be reversible phases of Li-rich compounds formed after Li
2SiO
3 lithiation, which have not yet been identified. Wang et al. claimed that Li
2SiO
3 reacts with Li
+ ions and generates Li
2O and Si
[32], but this claim needs further confirmation.
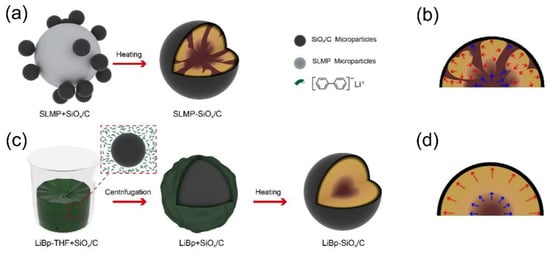
Figure 5. Schematic drawing illustrating the synthesis process and internal stresses for (
a,
b) SLMP-SiO
x/C and (
c,
d) LiBp-SiO
x/C. Reproduced with permission from ref.
[34]. Copyright 2020, ACS Publications.
Other than being generated in situ in electrochemical cells or via chemical lithiation processes with lithium metal, Li
2SiO
3 can be synthesized by using the hydrothermal method followed by a heat process
[29,31,32][29][31][32]. First, silicon and LiOH are well mixed in a Teflon-lined autoclave and heated at 180 °C for an appropriate dwell time, and then the product is transferred to a furnace under inert gas and sintered at 600 °C–800 °C; as a result, pure Li
2SiO
3 can be obtained
[29]. Hydrothermal synthesis using H
2SiO
3 as the starting material instead of silicon can also produce Li
2SiO
3 without post-heat treatments
[35]. A few studies have reported that Li
2SiO
3 and byproduct Li
2CO
3 can be formed when Li
4SiO
4 reacts with CO
2 gas
[28,30][28][30]. A water-based sol-gel method adopting fumed silica and LiOH can produce Li
2SiO
3 and Li
2CO
3 at 500 °C, but Li
4SiO
4 is the only dominant phase if the intermediate is heated up to 675 °C
[36]. Obviously, Li
2SiO
3 and Li
4SiO
4 are mutually convertible when Li
2CO
3 or CO
2 is introduced into the thermal process.
3.3. Li2Si2O5
Li
2Si
2O
5 was found as a result of characterizations of selected-area electron diffraction (SAED) under high-resolution transmission electron microscopy (HRTEM) with other lithium silicates (Li
4SiO
4 and Li
6Si
2O
7) in an NaOH-etched SiO anode during electrochemical lithiation and delithiation
[20]. All three lithium silicates were preserved, even when the Li metal half cell was discharged to 0 V
[19,20][19][20]. Li
2Si
2O
5 should be the lithium silicate phase formed in the early stage because of its low Li/Si ratio. However, only Li
2Si
2O
5 disappeared, while Li
4SiO
4 and Li
6Si
2O
7 remained after the cell was charged to 2 V. It was proposed that Li
4SiO
4 and Li
6Si
2O
7 are irreversible phases in LIBs, but Li
2Si
2O
5 can react with Si and then be converted into SiO
x during delithiation
[19,20,37][19][20][37] or react with additional Li
+ ions to obtain Li
2SiO
3 and Si
[15]. Another paper reporting a thin SiO
2 film electrode utilized in LIBs showed that the lithiated product Li
2Si
2O
5 has good cyclability and can be converted back to SiO
2 at 3.0 V vs. Li/Li
+ [38]. Lener et al. calculated the free energy for the lithiation reaction of silica and concluded that Li
2Si
2O
5 is, thermodynamically, the most favorable lithium silicate product
[39].
In addition to hydrothermal synthesis
[40], Li
2Si
2O
5 was able to be synthesized using carbon-coated SiO
x (SiO
x/C) microparticles and lithium-biphenyl (LiBp) complex solution, as shown in
Figure 5c. In contrast to the lithiation method utilizing stabilized lithium metal power (SLMP) exhibited in
Figure 5a, LiBp complex solution provides a homogeneous lithiation environment due to the intimate contact of SiO
x/C with the Li-containing solution. Chemical lithiation that adopts SLMP, usually in several microns, can only offer point-to-point lithiation channels where the particles are in contact with each other (
Figure 5a). The relatively inhomogeneous lithiation leads to randomly distributed Li
xSi
yO
z formation, resulting in strong internal stresses and thereby pulverization (
Figure 5b). The LiBp-SiO
x/C sample has a uniform Li
2Si
2O
5 coating on the particle; thereby, much better cycle performance and internal stress control can be obtained (
Figure 5d). Yan et al. attributed the formation of Li
2Si
2O
5 to the homogeneous absorption of LiBp on the SiO
x/C surface, which supports stable lithiation reactions, avoiding both excess and deficient lithium supply, as shown in the SLMP-SiO
x/C sample processed by solid-state lithiation
[34].
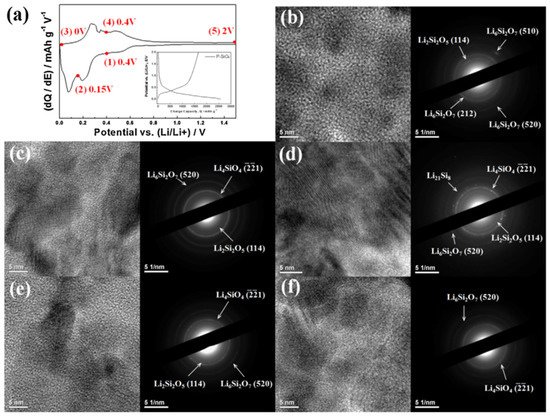
3.4. Li6Si2O7
As mentioned above, Li
6Si
2O
7 co-exists with Li
2Si
2O
5 and Li
4SiO
4 in SiO/SiO
X-based anodes during discharge and charge reactions
[19,20][19][20].
Figure 6 explicitly displays the generation and transformation of lithium silicate phases at different delithiation and lithiation voltages
[19]. At 0.4 V, Li
2Si
2O
5 and Li
6Si
2O
7 first appear in the anode, as shown in
Figure 6b. When further discharged to 0.15 V, the porous SiO
X anode starts to generate Li
4SiO
4, and both Li
2Si
2O
5 and Li
6Si
2O
7 remain in the anode structure until 0 V (
Figure 6c,d). All three lithium silicates can be still observed when the cell is charged to 0.4 V (
Figure 6e), which is a higher charge than that of the formation voltage of Li
4SiO
4. At a full charge of 2 V (exhibited in
Figure 6f) Li
2Si
2O
5 disappears, but Li
6Si
2O
7 and Li
4SiO
4 cannot be delithiated and remain in the structure as the dominant phases. Li
6Si
2O
7 is clearly an irreversible phase, like Li
4SiO
4, playing a role as a buffer material in SiO/SiO
X-based anodes to alleviate their swelling behavior. It is worth noting that a reversible Li
2SiO
3 phase was not detected in the reports from the same research group
[19[19][20][24],
20,24], in contrast to the results of a later study
[33]. A possible reason for this contradiction is that Li
2SiO
3 may be formed at a very early stage of lithiation and synchronously converted into Si and Li
2O due to further lithiation
[31,32][31][32].
Figure 6. (
a) Differential capacity plot (DCP) and charge/discharge profile of porous SiO
x anode at the first cycle. Ex situ HRTEM images with SAED patterns of porous SiO
x anode at various potentials indicated in DCP. (
b) Discharge at 0.4 V, (
c) discharge at 0.15 V, (
d) discharge at 0 V, (
e) charge at 0.4 V, and (
f) charge at 2 V. Reproduced with permission from ref.
[19]. Copyright 2013, Elsevier.
There are no studies in the literature reporting successful synthesis of sole Li
6Si
2O
7 as the only lithium silicate phase. In the previous studies, Li
6Si
2O
7 was synthesized, along with other lithium silicates, such as Li
2SiO
3 and Li
4SiO
4 [41,42][41][42]. Li
6Si
2O
7 (15%) was produced with Li
2SiO
3 (40%) and Li
4SiO
4 (45%) by annealing a melt consisting of 60Li
2O ⋅ 40SiO
2 prepared at 1250 °C
[41]. Mixed lithium silicate phases including Li
6Si
2O
7 as impurities were obtained in an LTAP (Li
1.
3Ti
1.7Al
0.3(PO
4)
3) sample with an Li
2O additive
[43]. Li
6Si
2O
7 as a minor phase can also be synthesized with Li
4SiO
4 using a sintering process of a Li
2CO
3 and SiO
2 mixture between 750 °C and 850 °C
[42]. However, the Li
6Si
2O
7 phase vanishes when the heating temperature is increased to 900 °C. It was reported that Li
6Si
2O
7 is a metastable lithium silicate phase, which can be decomposed into Li
2SiO
3 and Li
4SiO
4 at a high temperature
[44].
4. Lithium Silicates in Silicon, Lithium Metal, and Other Anode Materials
The success of lithium silicates in SiO/SiO
X-based anodes has prompted researchers to investigate their use in high-capacity silicon and lithium metal anode systems. Both anodes have unsettled problems, such as dramatic volume change, unstable interfacial reactions, and poor cycle performance
[45,46,47,48,49][45][46][47][48][49]. In a typical SiO anode, the lithium silicates formed in situ after the initial lithiation act as a rigid framework to accommodate the swelled Li
xSi alloy, and the lithium silicates in the structure can provide good ionic channels at the same time
[11].
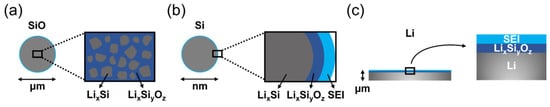
Figure 7 displays the common positional design of lithium silicates in SiO/SiO
x, nano-Si, and lithium metal anode materials. Both SiO and nano-Si are in powder form, but only SiO possesses a structure with dispersed lithium silicates. On the other hand, lithium silicates are usually applied as a coating layer in nano-Si and metallic lithium anodes. Methods of applying lithium silicates in anode structures have been extensively developed. Below,
we give some examples of how to employ lithium silicates in order to stabilize the electrochemical reactions of silicon and lithium metal anode materials, thereby prolonging their cycle lif
ee are given.
Figure 7. Schematic drawing illustrating the lithium silicates (LixSiyOz) in the structure of (a) SiO/SiOx, (b) nano-Si, and (c) metallic lithium anode materials.
4.1. Lithium Silicates in Silicon Anodes
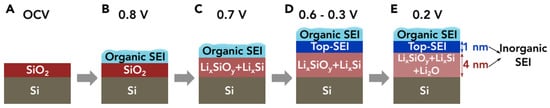
Cao et al. analyzed the composition of SEI formed on the native oxide-covered silicon anodes by employing in situ synchrotron X-ray reflectivity, linear sweep voltammetry, and ex situ XPS
[16]. The inorganic SEI comprised two well-defined structures, which can be divided into a bottom SEI layer (adjacent to the electrode) and a top SEI layer (adjacent to the organic SEI layer from electrolyte decomposition), as shown in
Figure 8. At open circuit voltage (OCV), the silicon surface is terminated by the pristine native silica (
Figure 8A). While lithiation begins and the anode voltage drops to 0.8 V vs. Li/Li
+, the organic SEI starts to form on the silicon anode, and native silica remains unchanged (
Figure 8B). At 0.7 V, the lithiation of the native oxide layer initiates and transforms into lithium silicates (Li
xSiO
y)
[16,50,51][16][50][51] and lithium silicide alloys (Li
xSi) as the bottom SEI layer (
Figure 8C). This finding is also in consistent with another report, which concluded that the lithiation voltage of SiO
2 is higher than that of Si
[14]. Between 0.6 V and 0.3 V, the top SEI, consisting of LiF and other products, is formed in between the organic SEI and bottom SEI, and lithiation of the bottom SEI continues (
Figure 8D). At 0.2 V, the last step of SEI formation, Li
2O is formed in the bottom SEI layer, which is thicker than the top SEI (
Figure 8E). The authors concluded that a smooth Li
xSiO
y layer as the bottom SEI can secure the growth of a conformal, smooth, and thin top SEI layer, and both cycle stability and rate performance can be guaranteed
[16]. It was reported that Li
4SiO
4 should be the final lithium silicate product from the lithiation of either native oxide or artificial oxide grown on an Si surface, and a small lithiation current is necessary to activate the electrochemical synthesis of lithium silicates
[23,52][23][52].
Figure 8. (
A–
E) Schematic drawing illustrating the proposed voltage-dependent SEI growth mechanism on native oxide-terminated silicon anodes. Reproduced with permission from ref.
[16]. Copyright 2018, Elsevier.
In addition to lithium-silicate-based SEI derived from native oxide, another approach to directly deposit lithium silicates onto silicon as an artificial SEI can be implemented for superior battery anode performance. Zhu et al. synthesized a Si@Li
2SiO
3 core-shell anode by oxidizing silicon nanoparticles at 600 °C in air with a subsequent thermal prelithiation process
[53]. In comparison with the non-prelithiated Si@SiO
x anode, Si@Li
2SiO
3 showed a much better initial Coulombic efficiency of 89.1% (vs. 66.5% in Si@SiO
x and 56.8% in Si), indicating prelithiation treatment can minimize irreversible Li-ion consumption during SEI formation reactions. Cycling tests also revealed that Si@Li
2SiO
3 electrodes outperformed Si@SiO
x and Si not only in specific capacity but also in cycling stability. The Li
2SiO
3 shell did slightly contribute to a charge/discharge capacity of 105.3/125.3 mA h g
−1, which is consistent with the results of lithium activity discussed in the previous section
[29]. The authors also demonstrated that the electron and Li-ion diffusion pathways of the Li
2SiO
3 layer on the Si surface are better than those of the SiO
x layer by comparing their reaction resistances, leading to an outstanding rate capability
[53].
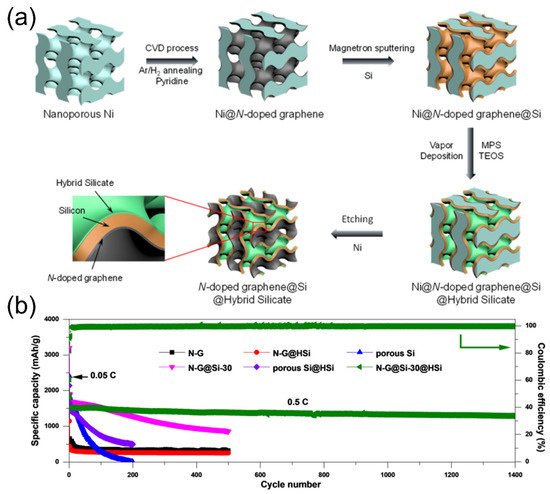
Huang et al. utilized nanoporous nickel as the substrate to be coated with N-doped graphene (via CVD process), silicon (via magnetron sputtering), and hybrid silicate (via vapor deposition) to build a sandwiched N-doped graphene@Si@hybrid silicate (N-G@Si@HSi) nanoarchitecture
[54].
Figure 9a shows the step-by-step details of the fabrication process of the bicontinuous porous material. A hybrid silicate was derived from the reactions of 3-mercaptopropyl trimethoxysilane (MPS), tetraethyl orthosilicate (TEOS) precursors, and H
2O vapors after a mild heating process. After the last nickel etching step, the free-standing film was used as the anode. The N-G@Si-30@HSi (Si-30: 59 nm silicon deposition thickness) exhibited extremely stable cycle performance among the other anode deigns in
Figure 9b. In the ingenious sandwiched anode design, the N-doped graphene serves as a flexible and conductive support, and the porous silicon nanolayer can avoid severe volume expansion and pulverization
[6,55,56,57,58,59,60,61][6][55][56][57][58][59][60][61]. The amorphous external coating of hybrid lithium silicates plays an important role to significantly enhance the robustness and integrity of the structure and to facilitate the formation of stable SEI films
[54].
Figure 9. (
a) Schematic drawing illustrating the fabrication process of the nanoporous N-G@Si@HSi anode. (
b) Cycling performance of the N-G, N-G@HSi, porous Si, N-G@Si-30, porous Si@HSi, and N-G@Si-30@HSi electrodes at 0.05 C for the initial three cycles and at 0.5 C for the extended cycles. Reproduced with permission from ref.
[54]. Copyright 2020, ACS Publications.
4.2. Lithium Silicates in Lithium Metal Anodes
A Li
4SiO
4 passivation layer can be formed on lithium metal anodes by in situ reactions between lithium foil and the a-SiO
2 thin film derived from electron cyclotron resonance (ECR)−chemical vapor deposition (CVD)
[62].
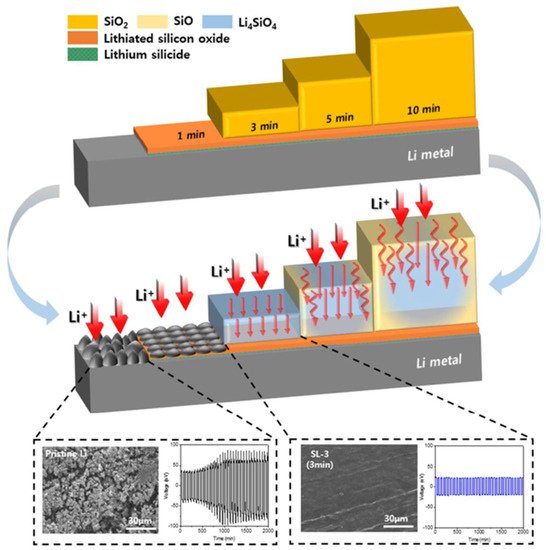
Figure 10 exhibits the hierarchical structure of the composite lithium metal anode, which varies with a-SiO
2 deposition time, which is proportional to the thickness of the passivation layer. Without any interfacial protection, a rough surface was clearly found in the pristine lithium anode after lithiation (bottom left in
Figure 10), and severe voltage polarization worsened with increasing cycle numbers due to significant degradation of the ether-based electrolyte
[63]. While a-SiO
2 was deposited onto the lithium metal, a thin lithium silicide and lithiated silicon oxide bilayer was simultaneously generated by the immediate reactions with lithium. With a longer deposition time, pure a-SiO
2 started to exist on top of the bilayer, and the thickness of a-SiO
2 grew with the process time. After lithiation, the a-SiO
2 top layer was converted into Li
4SiO
4, leading to a dendrite-free surface (SL-3 sample shown in the bottom of
Figure 10), showing a flat plating/stripping voltage plateau with a minimal overpotential of 21.7 mV during repeated cycling, which represents excellent interfacial stability
[62,64][62][64]. When the a-SiO
2 layer became too thick, not only ionically conductive Li
4SiO
4 but also relatively insulating SiO was formed in the artificial top-layer structure, resulting in a higher overpotential due to the thicker protective layer’s longer Li-ion migration distance and poorer diffusivity due to a higher SiO/Li
4SiO
4 area ratio
[62]. A few cracks were found in the lithium metal anode samples, particularly with a thicker a-SiO
2 protective layer, due to the heavy electrical stress induced by high electrical resistance, thereby leading to high interfacial resistance
[62,65][62][65]. As a result, owing to higher Li-ion conductivity and Young’s modulus (141.1 GPa) of Li
4SiO
4 than those of a-SiO
2, a carefully designed Li
4SiO
4 artificial layer can serve as an excellent protective interface to stabilize the electrochemical reactions of lithium metal anodes
[62].
Figure 10. Schematic drawing illustrating the silicon oxide-based passivation layer deposited on the surface of lithium metal anode by the ECR-CVD method. Reproduced with permission from ref.
[62]. Copyright 2018, ACS Publications.
A similar approach was developed by applying a-SiO
2 nanoparticles top of a lithium metal anode
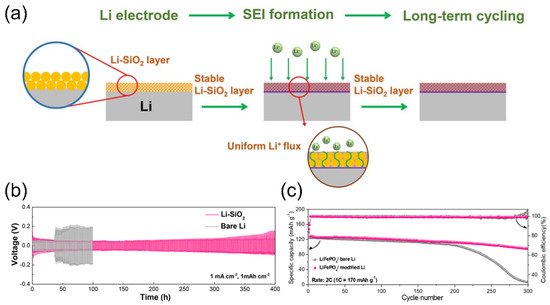
[66]. Silica spherical powders (200–300 nm in diameter) were sprayed onto a fresh lithium foil, followed by rolling-pressing to improve adherence. A compact and conformal SiO
2 layer with a thickness of 3.6 μm then functioned as the artificial interfacial layer to stabilize the lithium-plating and stripping behaviors in lithium metal batteries (
Figure 11a). The porous SiO
2 layer transformed into Li
4SiO
4 after prelithiation, and zigzagged channels that can facilitate Li-ion transfer were formed between Li
4SiO
4 particles. This protective film remained integrated after extended cycles, which guarantees low polarization (
Figure 11b) and great cycle performance (
Figure 11c). Unlike previously mentioned continuous a-SiO
2 film fabricated via the ECR-CVD method
[62], a layer consisting of a-SiO
2 powder has plenty of interstices, where SEI species, such as LiF, can infiltrate during initial formation cycles
[66]. This Li
4SiO
4 hybrid structure possesses higher Young’s modulus (up to 8.81 GPa), lower Li-ion diffusion barrier, and flexibility to accommodate volume expansion
[66]. Therefore, great cyclability and rate performance originating from stable interfacial reactions and enhanced charge-transfer kinetics can be obtained.
Figure 11. (
a) Schematic drawing illustrating the fabrication of a conformal Li
4SiO
4-based hybrid layer for the dendrite-proof metallic lithium anode. (
b) Electrochemical cycling stability of bare Li and modified Li symmetric cells (1 mA cm
−2, 1 mA h cm
−2). (
c) Long-term cycling performance of Li metal full cells. Reproduced with permission from ref.
[66]. Copyright 2020, ACS Publications.
Liu et al. directly exposed a piece of lithium foil to MPS and TEOS vapors, and lithium silicates (Li
xSi
yO
z) were formed and bound with surface Li
2O and LiOH, which were naturally generated on the lithium metal before vapor deposition
[67]. It was reported that when the hydroxyl group on lithium reacts with TEOS, the protective film formed in situ possesses a self-healing feature that can dynamically repair the corroded metallic lithium anode, which, remarkably, improves its reversibility
[68]. Since MPS and TEOS are both organosilicon compounds, methoxysilane (–Si–OCH
3, from MPS) and ethoxysilane (–Si–OCH
2CH
3, from TEOS) can undergo hydrolysis and condensation reactions to create a hybrid organic–inorganic lithium silicate layer comprising a “soft” organic moiety (mercaptopropyl groups) and a “hard” inorganic moiety (lithium silicates)
[67]. The soft part in the artificial layer can improve flexibility and robustness, and the hard part can enhance Li-ion transport and block the growth of lithium dendrite. Both organic and inorganic structures synergistically stabilize anode reactions of lithium metal during charge and discharge, leading to great cycle life in Li–LiFePO
4 and Li–S full cells
[67]. Ju et al. adopted aluminum silicate fibers to protect lithium metal anodes, realizing dendrite-free lithium anode reactions and a fast Li-ion conducting interface
[69]. Inorganic species of lithium aluminates (Li
xAlO
y) and lithium silicates (Li
xSiO
y) were detected upon cycling, implying alternative metal silicates could also be used as precursors of artificial SEI for lithium metal anodes.
Liang et al. floated lithium metal foil on a solution of 3-glycidyloxypropyltrimethoxysilane (GPTMS) and LiOH, and Li
6Si
2O
7 particles were adaptively embedded in the Si–O–Si polymer as a protective dual-phase layer during the initial lithium-stripping/plating process
[70]. This dual-phase interface showed high elastic modulus, high mechanical rigidity, and high interface energy, which can suppress the formation of one-dimensional lithium dendrite whiskers
[71]. Moreover, the structure of the composite shielding layer can efficiently lower the Li-ion migration barrier
[72]. An Li–air battery with an adaptive polymer-inorganic hybrid structure on the lithium metal anode can perform up to 180 cycles in an air atmosphere (25 °C, 15% humidity) with a capacity limitation of 1000 mAh g
−1 under a high current density of 1000 mA g
−1 [70]. Yu et al. employed the self-healing protective film concept by not only treating lithium metal foil in TEOS as the anode but using TEOS as an additive in the electrolyte for film-forming purposes during cycling
[68]. A thin lithium silicate layer can be formed under the reaction between TEOS and the detrimental corrosion products LiOH on the surface of lithium metal anodes in the highly corrosive environment of Li–O
2 batteries. The shielded lithium metal anode with film-forming electrolyte additive gives Li–O
2 batteries a considerable boost in cycle life (up to 144 cycles), indicating great dynamic-repair capability of the protective layer on the lithium metal anode
[68].
Lithium silicates can also be used to serve as a bridge layer between lithium metal anodes and solid-state electrolytes to lower the interfacial resistance. To achieve a low interfacial resistance with lithium metal of the garnet solid electrolyte, an amorphous Li
2SiO
3 interlayer was proposed
[73]. A liquid-phase coating process was utilized to deposit a thin lithium silicate film on the surface of garnet solid electrolytes, followed by a heat treatment, and metallic lithium was directly pressed onto the modified surface. This approach achieved stable cycling performance and a low interfacial resistance of 44 Ω cm
2, which was more than 10 times lower than that of the uncoated solid electrolyte. The authors proposed a possible reaction mechanism whereby Li
2SiO
3 decomposed into Li
21Si
5 and Li
2O after being in contact with lithium metal
[73].
To sum up, there are many approaches using lithium silicates with various Li/Si ratios to improve anode performance in Li-ion and Li metal batteries. The majority of previous research focused on the Li
4SiO
4 protective coating on silicon and metallic lithium anodes. With a rational design of composite anode structure, unwanted interfacial reactions can be either suppressed or eliminated, leading to better performance of long cycle life and small overpotential. The swelling issue of Si-based anodes can be addressed by utilizing SiO anodes where lithium silicates are formed in situ during cell formation or by adopting a 3D framework to accommodate lithium silicate-protected silicon.
Table 1 summarizes the previous works employing lithium silicates to boost battery anode performance. The studies related to modified metallic lithium anodes utilized excess lithium in the full cell. Without detailed full-cell design parameters, their specific capacity cannot be confirmed at this point. Still, other lithium silicate phases, such as Li
2Si
2O
5 and Li
6Si
2O
7, have yet to be identified as a good anode-protection-layer material, like Li
4SiO
4 and Li
2SiO
3. As a result, additional in-depth research is required before a sound judgment can be reached.
Table 1. Electrochemical performance of anode materials protected by lithium silicates.
5. Summary and Outlook
The properties of various lithium silicate phases have been carefully reviewed in this paper. Li
4SiO
4, regarded as the major compound among lithiated SiO anodes and SEI deposited on the naturally oxidized surface of Si anodes, is the most investigated lithium silicate material in battery applications. Li
4SiO
4 and Li
2SiO
3 are frequently used to protect high-capacity silicon and lithium metal anodes. Li
2Si
2O
5 and Li
6Si
2O
7 can also be found in some SiO
x-based battery anodes after the lithiation process or during charge/discharge reactions. In general, lithium silicates play an important role in anodes for (1) stabilization of interfacial reactions, (2) buffering of volume expansion without any mechanical failure, and (3) offering fast Li-ion transportation pathways. Further systematic research is required to determine which lithium silicate phase is the best in class to protect silicon and lithium anode materials.
Lithium silicates can be synthesized by lithiation of amorphous silica or organosilicon compounds. The necessary raw materials are commonly seen in chemical engineering and semiconductor industries and are low-cost and easy to process, making this approach feasible and scalable for battery anode applications. A streamlined manufacturing process needs to be developed for better integration into current battery-production procedures. Aside from the success of SiO/SiO
x anode materials, lithium silicates have now been successfully adopted in high-capacity anode systems, especially serving as the major ingredient in the protection layer of metallic lithium. Alternative lithium metal oxide compounds could be developed if comparable ionic and mechanical properties can be obtained. Due to the rigid nature of ceramics-like oxide-based ion-conducting materials that might lose intimate contact with the active anode material during cycling, a polymer-inorganic (soft-rigid) hybrid protection layer appears to be a promising option for achieving both high elasticity and ionic conductivity to shield silicon and lithium metal anodes
[67,70][67][70].
Currently, the lithium silicates adopted in commercial battery systems are mostly formed during the lithiation process of SiO anodes at the formation step of battery-cell manufacturing. The challenges of lithium silicate-enhanced anodes are: (1) The efficacy of anode protection by lithium silicates synthesized from silica or other organosilicon compounds other than SiO is still not clear at the industry level; (2) The processing and preparation of lithium silicate protective layers could be cost-ineffective; (3) The long-term protection and adhesion stability of lithium silicates in integrated anodes is unknown. Strategies concerning smart architectural designs, innovative methods of anode integration processes, low-cost and scalable synthesis, and long-cycle durability improvements will need to be thoroughly investigated before lithium silicates can be widely applied in various anode systems for practical Li-ion and Li metal batteries.