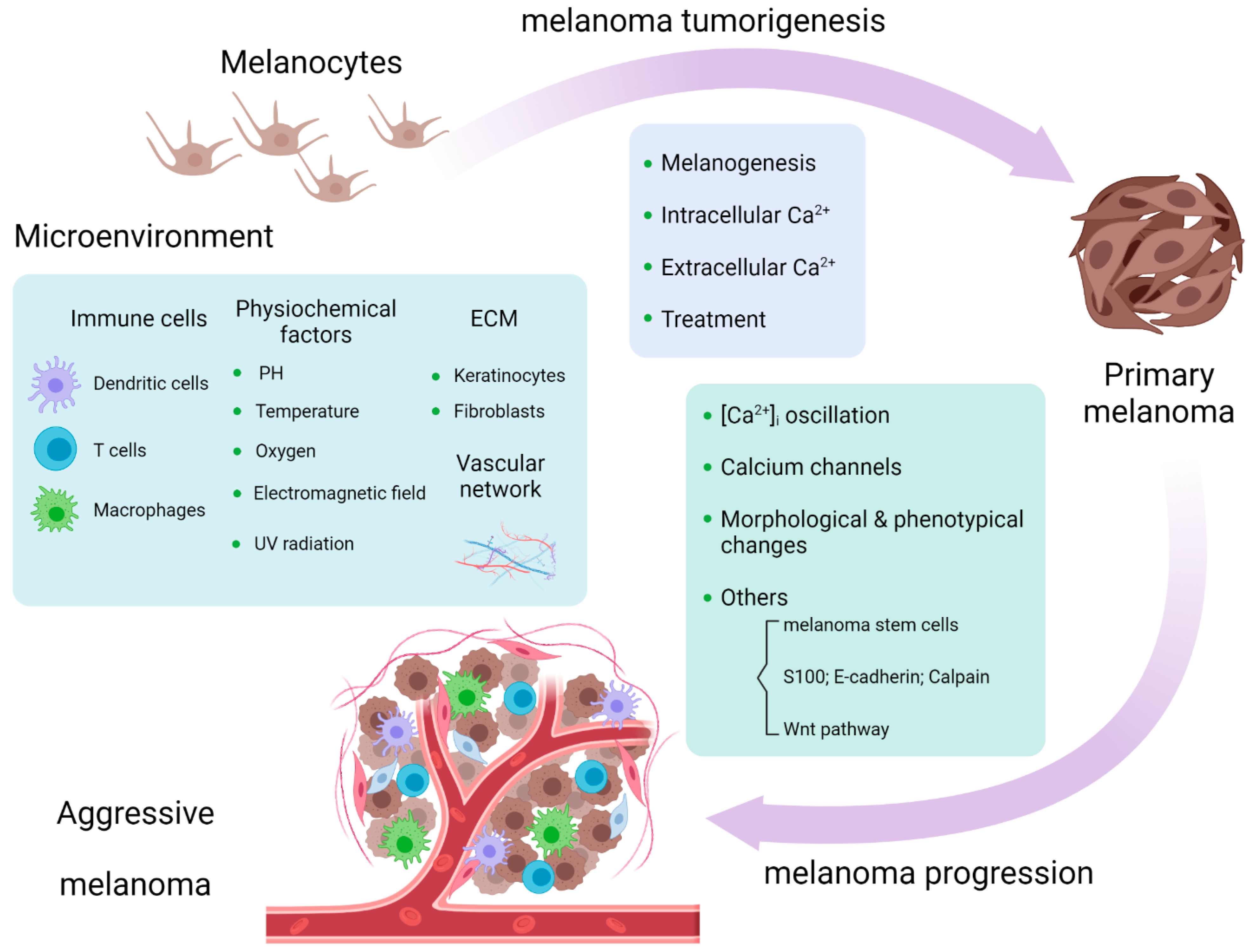
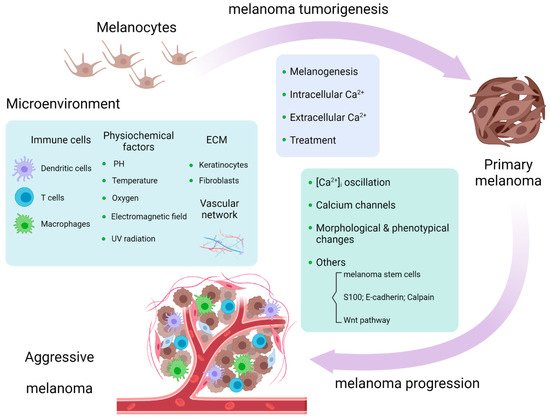
Since [Ca
2+]i plays an important mechanistic role in melanoma progression, the role of calcium channels cannot be neglected. Basically, NMDAR calcium channel function is weak in melanoma cells but strongly contributes to cell proliferation and invasion when its encoding gene GRIN2A is mutated at certain sites, such as G762E, with less glutamate supplementation
[7][34]. Another glutamate receptor calcium channel mGluR5 was proved to have a profound effect on melanoma progression in vivo by triggering the phosphorylation of ERK
[8][5]. The ERK pathway is also implicated in SOCE-mediated melanoma progression. Inhibition of SOCE by knockdown of STIM1 or Orai or by SOCE inhibitors suppresses melanoma cell proliferation and migration, while induction of SOCE activates ERK, which is inhibited by calmodulin kinase II or Raf-1 inhibitors
[9][35]. TPC2 influences melanoma progression via SOCE. Downregulation of TPC2 expression in metastatic melanoma leads to a decrease of Orai1 expression and an increase of YAP/TAZ activity, which is responsible for melanoma’s aggressive property
[10][36]. In BRAF mutant melanoma—the BRAF
V600E mutation in particular—the expression of Ca
2+-ATPase isoform 4b (PMCA4b) on the plasma membrane is low compared with benign nevi and is markedly elevated by vemurafenib (BRAF inhibitor) or selumetinib (MEK inhibitor) treatment, which indicates crosstalk between PMCA4b and the MAPK pathway. Activation of p38 MAPK induces the degradation of PMCA4b, while suppression of p38 MAPK by increasing the abundance of PMCA4b promotes the [Ca
2+]i clearance and inhibits the migration of melanoma cells
[11][12][37,38]. Moreover, SERCA on the ER membrane, controlled by the interaction between calcium-modulating cyclophilin ligand and basigin, was reported to have an effect on invasion and metastasis by regulating [Ca
2+]i and matrix metalloproteinase (MMP)-9 activity in A375 cells
[13][39]. Unlike Ca
2+-ATPase, T-type VDCCs drive migration and invasion in BRAF mutant melanoma cells depending on Snail1 levels, suggesting therapeutic strategies by blocking T-type VDCCs to inhibit progression of melanoma
[14][40]. Other ion channels are implicated in melanoma progression through calcium signaling. Nav1.6 sodium channel promotes melanoma cell (WM266 and WM115) invasion and proliferation by mTOR-mediated Na
+/Ca2
+ exchange
[15][41]. KCa3.1 potassium channel was reported to promote melanoma cell migration by controlling the secretion of melanoma inhibitory activity proteins depending on [Ca
2+]i
[16][42].