Non-alcoholic fatty liver disease (NAFLD) is a slowly progressing disease, beginning with isolated liver steatosis that evolves in a subset of patients to non-alcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC). It was recently proposed to redefine NAFLD to metabolic dysfunction-associated fatty liver disease (MAFLD) in which other known causes of liver disease such as alcohol consumption or viral hepatitis do not need to be excluded. Revised nomenclature envisions speeding up and facilitating anti-MAFLD drug development by means of patient stratification whereby each subgroup would benefit from distinct pharmacological interventions.
- NAFLD
- MAFLD
- NASH
- liver
- drug development
- pharmacology
- in vitro
1. Introduction
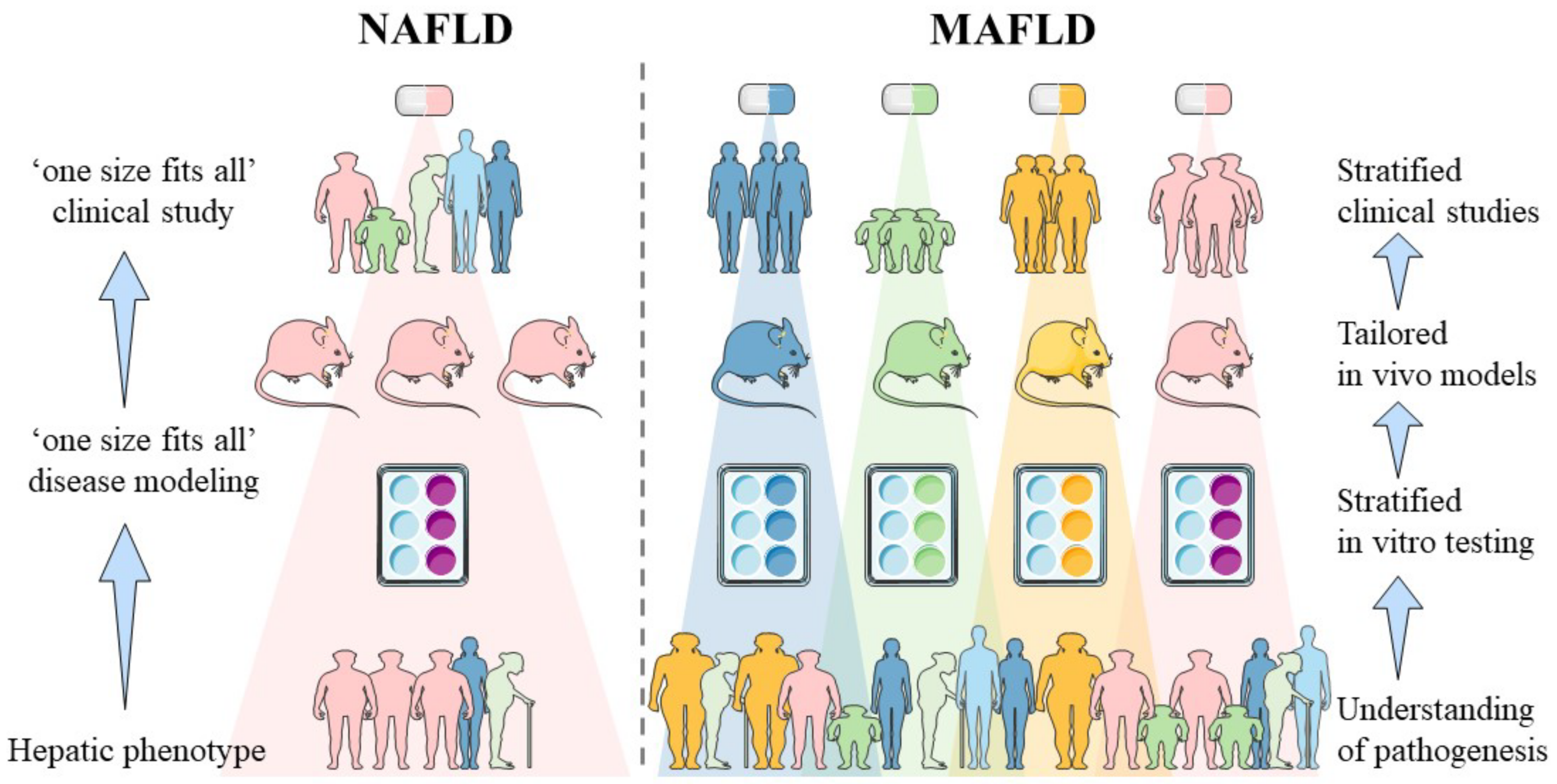
2. Sources of Heterogeneity in MAFLD and Their In Vitro Implementation
2.1. Sex and Hormonal Status
2.2. Pediatric/Juvenile MAFLD
2.3. Fructose Consumption
2.4. Genetic Predisposition and Ethnicity
2.5. Epigenetics
2.6. Obesity and Body Fat Distribution
2.7. Lean MAFLD
2.8. Microbiota
2.9. Zonation
2.10. Disease Progression and Regression
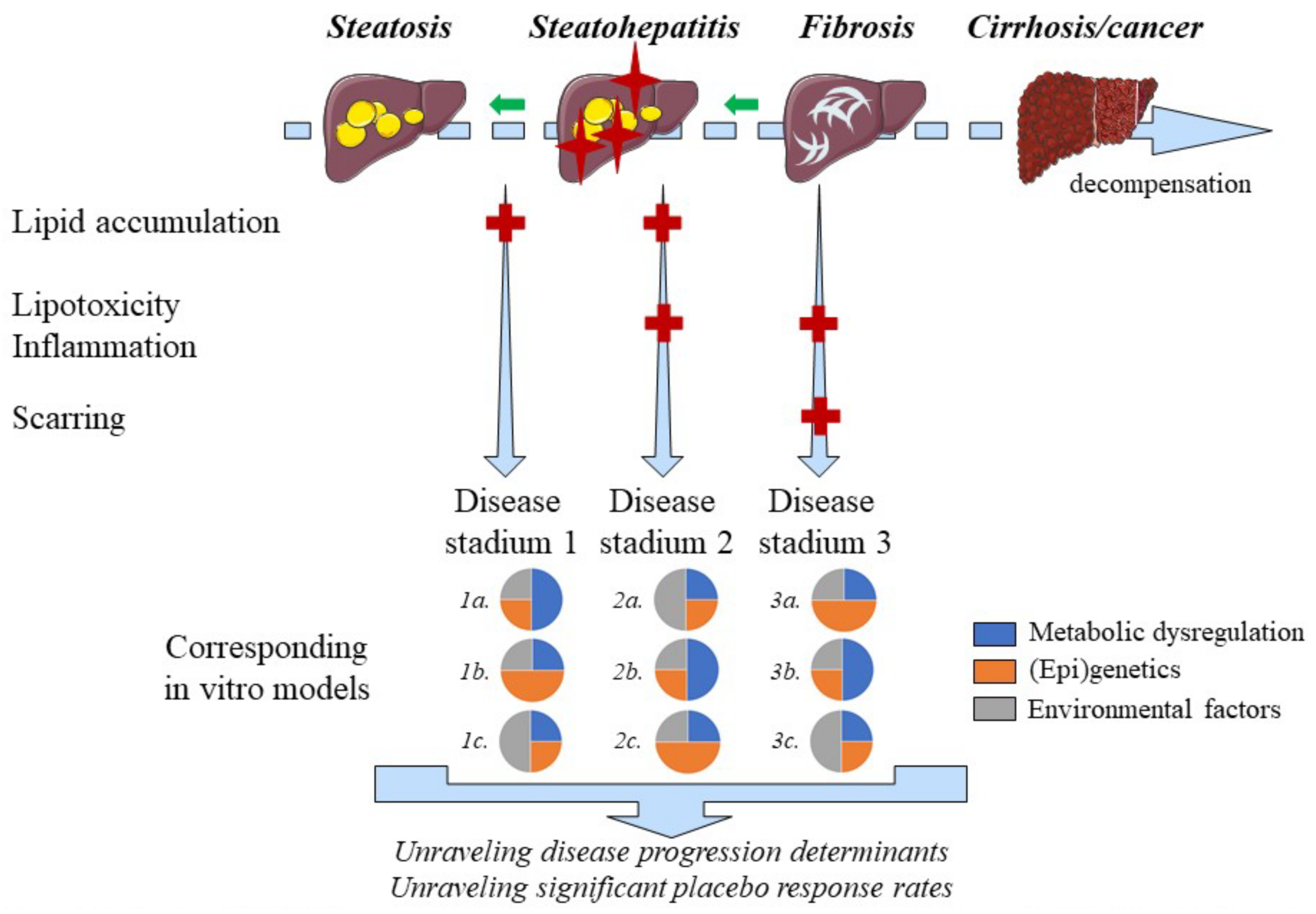
2.11. Ethanol consumption
Conflicting data exists regarding the effects of ethanol consumption in patients suffering from NMAFLD. A prospective cohort study including 4568 NAFLD patients concluded that modest ethanol consumption (i.e., 0.5 to 1.5 standard drinks per day) decreases all-cause mortality compared to ethanol abstinence. In contrast, drinking more than 1.5 standard drinks of ethanol per day, rapidly increases all-cause mortality [125][92]. These findings are consistent with the fact that moderate ethanol intake reduces the risk for several cardiovascular outcomes [126][93] and that the beneficial or detrimental effects of ethanol on different health aspects follow a ‘J-shaped’ curve [125][92]. Prohibiting ethanol consumption in clinical studies is far from practical, often does not reflect the daily situation and would narrow the definition of MAFLD [7,127][7][94]. Therefore, pharmacological effects of novel anti-NASH compounds could be evaluated in vitro with different alcohol concentrations to gain insights in situations that reflect daily life for many patients. Ethanol induces transcriptional activity of SREBP-1c and carbohydrate-responsive element-binding protein thereby promoting lipogenesis [128][95]. SREBP-1c levels, however, decrease in patients with NASH progression [129][96]. Concomitant exposure of FaO hepatoma cells to ethanol and free fatty acids reduces lipid droplet size compared to single treatments [130],[97], which correlates with more advanced MAFLD histology [131][98]. Coexisting alcoholic- and non-alcoholic fatty liver disease might therefore be an important but underrecognized disease. Evaluation of compounds using in vitro systems, in which both the effects of ethanol and metabolic factors are modeled, can facilitate the development of drugs targeting mixed pathologies.
References
- Cotter, T.G.; Rinella, M. Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology 2020, 158, 1851–1864.
- Boeckmans, J.; Natale, A.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Human-based systems: Mechanistic NASH modelling just around the corner? Pharmacol. Res. 2018, 134, 257–267.
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; Lavine, J.E.; Ratziu, V.; Mccullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011, 54, 344–353.
- Cohen, D.E.; Fisher, E.A. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin. Liver Dis. 2013, 33, 380–388.
- Corey, K.E.; Misdraji, J.; Gelrud, L.; Zheng, H.; Chung, R.T.; Krauss, R.M. Nonalcoholic steatohepatitis is associated with an atherogenic lipoprotein subfraction profile. Lipids Health Dis. 2014, 13, 100.
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH drug development hitches a lift on PPAR agonism. Cells 2020, 9, 37.
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H.; et al. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014.
- Han, M.A.T.; Altayar, O.; Hamdeh, S.; Takyar, V.; Rotman, Y.; Etzion, O.; Lefebvre, E.; Safadi, R.; Ratziu, V.; Prokop, L.J.; et al. Rates of and factors associated with placebo response in trials of pharmacotherapies for nonalcoholic steatohepatitis: Systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 616–629.e26.
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015, 149, 367–378.
- Glass, O.; Filozof, C.; Noureddin, M.; Berner-Hansen, M.; Schabel, E.; Omokaro, S.O.; Schattenberg, J.M.; Barradas, K.; Miller, V.; Francque, S.; et al. Standardisation of diet and exercise in clinical trials of NAFLD-NASH: Recommendations from the Liver Forum. J. Hepatol. 2020, 73, 680–693.
- Abdelmalek, M.F. Nonalcoholic fatty liver disease: Another leap forward. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 85–86.
- Huang, J.; Kumar, R.; Wang, M.; Zhu, Y.; Lin, S. MAFLD criteria overlooks a number of patients with severe steatosis: Is it clinically relevant? J. Hepatol. 2020, 73, 1265–1267.
- Ciardullo, S.; Perseghin, G. Prevalence of NAFLD, MAFLD and associated advanced fibrosis in the contemporary United States population. Liver Int. 2021, 41, 1290–1293.
- Chella Krishnan, K.; Floyd, R.R.; Sabir, S.; Jayasekera, D.W.; Leon-Mimila, P.V.; Jones, A.E.; Cortez, A.A.; Shravah, V.; Péterfy, M.; Stiles, L.; et al. Liver pyruvate kinase promotes NAFLD/NASH in both mice and humans in a sex-specific manner. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 389–406.
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131.
- Vandel, J.; Dubois-Chevalier, J.; Gheeraert, C.; Derudas, B.; Raverdy, V.; Thuillier, D.; Gaal, L.; Francque, S.; Pattou, F.; Staels, B.; et al. Hepatic molecular signatures highlight the sexual dimorphism of nonalcoholic steatohepatitis (NASH). Hepatology 2021, 73, 920–936.
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767.
- Shadid, S.; Abosi-Appeadu, K.; De Maertelaere, A.S.; Defreyne, J.; Veldeman, L.; Holst, J.J.; Lapauw, B.; Vilsbøll, T.; T’Sjoen, G. Effects of gender-affirming hormone therapy on insulin sensitivity and incretin responses in transgender people. Diabetes Care 2020, 43, 411–417.
- Shen, M.; Shi, H. Sex hormones and their receptors regulate liver energy homeostasis. Int. J. Endocrinol. 2015, 2015, 294278.
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649.
- Smith, S.K.; Perito, E.R. Nonalcoholic liver disease in children and adolescents. Clin. Liver Dis. 2018, 22, 723–733.
- Nobili, V.; Mosca, A.; De Vito, R.; Raponi, M.; Scorletti, E.; Byrne, C.D. Liver zonation in children with non-alcoholic fatty liver disease: Associations with dietary fructose and uric acid concentrations. Liver Int. 2018, 38, 1102–1109.
- Africa, J.A.; Behling, C.A.; Brunt, E.M.; Zhang, N.; Luo, Y.; Wells, A.; Hou, J.; Belt, P.H.; Kohil, R.; Lavine, J.E.; et al. In children with nonalcoholic fatty liver disease, zone 1 steatosis is associated with advanced fibrosis. Clin. Gastroenterol. Hepatol. 2018, 16, 438–446.
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.T.; Lawlor, D.A.; Fraser, A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0140908.
- Doycheva, I.; Watt, K.D.; Alkhouri, N. Nonalcoholic fatty liver disease in adolescents and young adults: The next frontier in the epidemic. Hepatology 2017, 65, 2100–2109.
- Zhang, X.; Wu, M.; Liu, Z.; Yuan, H.; Wu, X.; Shi, T.; Chen, X.; Zhang, T. Increasing prevalence of NAFLD/NASH among children, adolescents and young adults from 1990 to 2017: A population-based observational study. BMJ Open 2021, 11, 42843.
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228.
- Mulder, J.; Sharmin, S.; Chow, T.; Rodrigues, D.C.; Hildebrandt, M.R.; D’Cruz, R.; Rogers, I.; Ellis, J.; Rosenblum, N.D. Generation of infant- and pediatric-derived urinary induced pluripotent stem cells competent to form kidney organoids. Pediatr. Res. 2020, 87, 647–655.
- Assy, N.; Nasser, G.; Kamayse, I.; Nseir, W.; Beniashvili, Z.; Djibre, A.; Grosovski, M. Soft drink consumation linked with fatty liver in the absence of traditional risk factors. Can. J. Gastroenterol. 2008, 22, 811–816.
- Abid, A.; Taha, O.; Nseir, W.; Farah, R.; Grosovski, M.; Assy, N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J. Hepatol. 2009, 51, 918–924.
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999.
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, 625–631.
- Tappy, L.; Rosset, R. Health outcomes of a high fructose intake: The importance of physical activity. J. Physiol. 2019, 597, 3561–3571.
- Steinmann, B.; Ranter, R. Disorders of fructose metabolism. In Inborn Metabolic Diseases; Saudubray, J.M., van den Berghe, G., Walter, J.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 158–165.
- Shepherd, E.L.; Saborano, R.; Northall, E.; Matsuda, K.; Ogino, H.; Yashiro, H.; Pickens, J.; Feaver, R.E.; Cole, B.K.; Hoang, S.A.; et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep. 2021, 3, 100217.
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045.
- Samji, N.S.; Snell, P.D.; Singal, A.K.; Satapathy, S.K. Racial disparities in diagnosis and prognosis of nonalcoholic fatty liver disease. Clin. Liver Dis. 2020, 16, 66–72.
- Rich, N.E.; Oji, S.; Mufti, A.R.; Browning, J.D.; Parikh, N.D.; Odewole, M.; Mayo, H.; Singal, A.G. Racial and ethnic disparities in non-alcoholic fatty liver disease prevalence, severity, and outcomes in the United States: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 198–210.
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279.
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016, 150, 1219–1230.
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356.
- Liu, J.; Perry, R.C.; Ngai, C.; Ginsberg, H.N. Abstract 682: TM6SF2 is necessary for the late addition of lipid and secretion of fully-lipidated very low density lipoproteins from Hepg2 cell. Arterioscler. Thromb. Vasc. Biol. 2018, 36, A682.
- Borén, J.; Adiels, M.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Ståhlman, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of TM6SF2 E167K on hepatic lipid and very low-density lipoprotein metabolism in humans. JCI Insight 2020, 5, e144079.
- Luukkonen, P.K.; Nick, A.; Hölttä-Vuori, M.; Thiele, C.; Isokuortti, E.; Lallukka-Brück, S.; Zhou, Y.; Hakkarainen, A.; Lundbom, N.; Peltonen, M.; et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019, 4, e127902.
- Bruschi, F.V.; Claudel, T.; Caligiuri, A.; Marra, F.; Trauner, M.H. The I148M PNPLA3 variant is a novel key player modulating the pro-fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890.
- Tilson, S.G.; Morell, C.M.; Lenaerts, A.-S.; Park, S.B.; Hu, Z.; Jenkins, B.; Koulman, A.; Liang, T.J.; Vallier, L. Modeling PNPLA3-associated NAFLD using human-induced pluripotent stem cells. Hepatology 2021, 74, 2998–3017.
- Graffmann, N.; Ncube, A.; Martins, S.; Fiszl, A.R.; Reuther, P.; Bohndorf, M.; Wruck, W.; Beller, M.; Czekelius, C.; Adjaye, J. A stem cell based in vitro model of NAFLD enables the analysis of patient specific individual metabolic adaptations in response to a high fat diet and AdipoRon interference. Biol. Open 2021, 10, bio054189.
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484.
- Pirola, C.J.; Gianotti, T.F.; Castaño, G.O.; Mallardi, P.; San Martino, J.; Ledesma, M.M.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812.
- He, Y.; Hwang, S.; Cai, Y.; Kim, S.J.; Xu, M.; Yang, D.; Guillot, A.; Feng, D.; Seo, W.; Hou, X.; et al. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology 2019, 70, 1150–1167.
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2012, 62, 1356–1363.
- Page, A.; Pauli, P.; Morán-Salvador, E.; White, S.; French, J.; Mann, J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. Physiol. Behav. 2017, 64, 661–673.
- Tian, Y.; Wong, V.W.-S.; Chan, H.L.-Y.; Cheng, A.S.-L. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin. Cancer Biol. 2013, 23, 471–482.
- Dreval, K.; Tryndyak, V.; de Conti, A.; Beland, F.A.; Pogribny, I.P. Gene expression and DNA methylation alterations during nonalcoholic steatohepatitis-associated liver carcinogenesis. Front. Genet. 2019, 10, 486.
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84.
- van Vliet-Ostaptchouk, J.V.; Nuotio, M.L.; Slagter, S.N.; Doiron, D.; Fischer, K.; Foco, L.; Gaye, A.; Gögele, M.; Heier, M.; Hiekkalinna, T.; et al. The prevalence of metabolic syndrome and metabolically healthy obesity in Europe: A collaborative analysis of ten large cohort studies. BMC Endocr. Disord. 2014, 14, 9.
- Blüher, M. Metabolically healthy obesity. Endocr. Rev. 2020, 41, bnaa004.
- Wang, B.; Zhuang, R.; Luo, X.; Yin, L.; Pang, C.; Feng, T.; You, H.; Zhai, Y.; Ren, Y.; Zhang, L.; et al. Prevalence of metabolically healthy obese and metabolically obese but normal weight in adults worldwide: A meta-analysis. Horm. Metab. Res. 2015, 47, 839–845.
- Zheng, Q.; Lin, W.; Liu, C.; Zhou, Y.; Chen, T.; Zhang, L.; Zhang, X.; Yu, S.; Wu, Q.; Jin, Z.; et al. Prevalence and epidemiological determinants of metabolically obese but normal-weight in Chinese population. BMC Public Health 2020, 20, 487.
- Kim, D.; Chung, G.E.; Kwak, M.S.; Seo, H.B.; Kang, J.H.; Kim, W.; Kim, Y.J.; Yoon, J.H.; Lee, H.S.; Kim, C.Y. Body fat distribution and risk of incident and regressed nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2016, 14, 132–138.
- van der Poorten, D.; Milner, K.-L.; Hui, J.; Hodge, A.; Trenell, M.I.; Kench, J.G.; London, R.; Peduto, T.; Chisholm, D.J.; George, J. Visceral fat: A key mediator of steatohepatitis in metabolic liver disease. Hepatology 2008, 48, 449–457.
- Yu, S.J.; Kim, W.; Kim, D.; Yoon, J.H.; Lee, K.; Kim, J.H.; Cho, E.J.; Lee, J.H.; Kim, H.Y.; Kim, Y.J.; et al. Visceral obesity predicts significant fibrosis in patients with nonalcoholic fatty liver disease. Medicine 2015, 94, e2159.
- Cho, S.A.; Joo, H.J.; Cho, J.Y.; Lee, S.H.; Park, J.H.; Hong, S.J.; Yu, C.W.; Lim, D.S. Visceral fat area and serum adiponectin level predict the development of metabolic syndrome in a community-based asymptomatic population. PLoS ONE 2017, 12, e0169289.
- Leite, N.C.; Salles, G.F.; Cardoso, C.R.L.; Villela-Nogueira, C.A. Serum biomarkers in type 2 diabetic patients with non-alcoholic steatohepatitis and advanced fibrosis. Hepatol. Res. 2013, 43, 508–515.
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846.
- Jonas, M.I.; Kurylowicz, A.; Bartoszewicz, Z.; Lisik, W.; Jonas, M.; Wierzbicki, Z.; Chmura, A.; Pruszczyk, P.; Puzianowska-Kuznicka, M. Interleukins 6 and 15 levels are higher in subcutaneous adipose tissue, but obesity is associated with their increased content in visceral fat depots. Int. J. Mol. Sci. 2015, 16, 25817–25830.
- Park, H.S.; Park, J.Y.; Yu, R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res. Clin. Pract. 2005, 69, 29–35.
- Wieckowska, A.; Papouchado, B.G.; Li, Z.Z.; Lopez, R.; Zein, N.N.; Feldstein, A.E. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2008, 103, 1372–1379.
- Lu, F.B.; Zheng, K.I.; Rios, R.S.; Targher, G.; Byrne, C.D.; Zheng, M.H. Global epidemiology of lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2020, 35, 2041–2050.
- Ayonrinde, O.T. Historical narrative from fatty liver in the nineteenth century to contemporary NAFLD—Reconciling the present with the past. JHEP Rep. 2021, 3, 100261.
- Chen, F.; Esmaili, S.; Rogers, G.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A distinct entity shaped by differential metabolic adaptation. Hepatology 2020, 71, 1213–1227.
- Fracanzani, A.L.; Petta, S.; Lombardi, R.; Pisano, G.; Russello, M.; Consonni, D.; Di Marco, V.; Cammà, C.; Mensi, L.; Dongiovanni, P.; et al. Liver and cardiovascular damage in patients with lean nonalcoholic fatty liver disease, and association with visceral obesity. Clin. Gastroenterol. Hepatol. 2017, 15, 1604–1611.
- Younes, R.; Govaere, O.; Petta, S.; Miele, L.; Tiniakos, D.; Burt, A.; David, E.; Vecchio, F.M.; Maggioni, M.; Cabibi, D.; et al. Caucasian lean subjects with non-alcoholic fatty liver disease share long-term prognosis of non-lean: Time for reappraisal of BMI-driven approach? Gut 2022, 71, 382–390.
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609.
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775.
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2015, 91, 1–9.
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.N.; Li, S.; Xue, G.; et al. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.
- Ohtani, N.; Kawada, N. Role of the gut-liver axis in liver inflammation, fibrosis, and cancer: A special focus on the gut microbiota relationship. Hepatol. Commun. 2019, 3, 456–470.
- Jeschke, M.G.; Klein, D.; Thasler, W.E.; Bolder, U.; Schlitt, H.J.; Jauch, K.W.; Weiss, T.S. Insulin decreases inflammatory signal transcription factor expression in primary human liver cells after LPS challenge. Mol. Med. 2008, 14, 11–19.
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; Stefanis, C.D.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 2015, 6, 41434–41452.
- Kheder, R.K.; Hobkirk, J.; Stover, C.M. In vitro modulation of the LPS-induced proinflammatory profile of hepatocytes and macrophages- approaches for intervention in obesity? Front. Cell Dev. Biol. 2016, 4, 61.
- Aragonès, G.; Colom-Pellicer, M.; Aguilar, C.; Guiu-Jurado, E.; Martínez, S.; Sabench, F.; Antonio Porras, J.; Riesco, D.; Del Castillo, D.; Richart, C.; et al. Circulating microbiota-derived metabolites: A “liquid biopsy? Int. J. Obes. 2020, 44, 875–885.
- Jungermann, K.; Kietzmann, T. Oxygen: Modulator of metabolic zonation and disease of the liver. Hepatology 2000, 31, 255–260.
- Hijmans, B.S.; Grefhorst, A.; Oosterveer, M.H.; Groen, A.K. Zonation of glucose and fatty acid metabolism in the liver: Mechanism and metabolic consequences. Biochimie 2014, 96, 121–129.
- Yeh, M.M.; Brunt, E.M. Pathological features of fatty liver disease. Gastroenterology 2014, 147, 754–764.
- Hall, Z.; Bond, N.J.; Ashmore, T.; Sanders, F.; Ament, Z.; Wang, X.; Murray, A.J.; Bellafante, E.; Virtue, S.; Vidal-Puig, A.; et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1165–1180.
- Lee-Montiel, F.T.; George, S.M.; Gough, A.H.; Sharma, A.D.; Wu, J.; DeBiasio, R.; Vernetti, L.A.; Taylor, D.L. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp. Biol. Med. 2017, 242, 1617–1632.
- Sullivan, M.; Galea, P.; Latif, S. What is the appropriate oxygen tension for in vitro culture? Mol. Hum. Reprod. 2006, 12, 653.
- Parafati, M.; Kirby, R.J.; Khorasanizadeh, S.; Rastinejad, F.; Malany, S. A nonalcoholic fatty liver disease model in human induced pluripotent stem cell-derived hepatocytes, created by endoplasmic reticulum stress-induced steatosis. Dis. Model. Mech. 2018, 11, dmm033530.
- Feaver, R.E.; Cole, B.K.; Lawson, M.J.; Hoang, S.A.; Marukian, S.; Blackman, B.R.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R.; Dash, A. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. J. Clin. Invest. 2016, 1, e90954.
- Hajifathalian, K.; Sagvand, B.T.; Mccullough, A.J. Effect of alcohol consumption on survival in non-alcoholic fatty liver disease: A national prospective cohort study. Hepatology 2019, 70, 511–521.
- Ronksley, P.E.; Brien, S.E.; Turner, B.J.; Mukamal, K.J.; Ghali, W.A. Association of alcohol consumption with selected cardiovascular disease outcomes: A systematic review and meta-analysis. BMJ 2011, 342, d671.
- Eslam, M.; Sanyal, A.J.; George, J. Toward more accurate nomenclature for fatty liver diseases. Gastroenterology 2019, 157, 590–593.
- You, M.; Arteel, G.E. Effect of ethanol on lipid metabolism. J. Hepatol. 2019, 70, 237–248.
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 2010, 53, 724–731.
- Vecchione, G.; Grasselli, E.; Compalati, A.D.; Ragazzoni, M.; Cortese, K.; Gallo, G.; Voci, A.; Vergani, L. Ethanol and fatty acids impair lipid homeostasis in an in vitro model of hepatic steatosis. Food Chem. Toxicol. 2016, 90, 84–94.
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659.