Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Irem Ozel and Version 2 by Bruce Ren.
Tumor angiogenesis is one of the most critical steps in the progression of cancer. Owing to its fundamental role in tumor growth and metastasis, tumor angiogenesis is accepted to be a limiting factor and considered a powerful therapeutic target. Neutrophils contribute to the tumor progression via multiple mechanisms, ranging from the direct support of tumor cell survival to the immunosuppression. A strong body of evidence suggests that neutrophils also play a prominent role in tumor angiogenesis.
- tumor vasculature
- neutrophils
- cancer
- tumor angiogenesis
1. Introduction
The functional vascular system, in complex a multicellular organism, is a very vital asset for survival. The network of blood vessels nurtures all the tissues by supplying oxygen and nutrients, as well as providing gateways for immune surveillance [1][2][1,2]. In the early development of the embryo, mesoderm-derived endothelial precursor cells (termed angioblasts) constituted vasculogenesis. In later stages of embryonic development, the tree-like structure of blood vessels were generated from this initial vasculature, in a process called “angiogenesis” [3]. Although the new vessel formation occurs mainly during the embryonic development, angiogenesis also takes place in the adult organism and plays a crucial role in physiological conditions, such as wound healing, placenta formation, and menstrual cycles [3]. Next to physiological angiogenesis, abnormal development of new blood vessels occurs in multiple pathological conditions, such as cancer. Tumor angiogenesis is essential for cancer progression because tumors cannot grow beyond 1 mm3 without sufficient blood supply [4]. Moreover, the spread of tumor cells is strongly dependent on functional vasculature [5]. Therefore, tumor cells release factors that modulate the activity of immune cells, including neutrophils, to support their pro-angiogenic activity.
2. Neutrophils in Cancer
Neutrophils, as the most abundant leukocytes in human blood circulation, ranging between 50 and 70 percent (10–25 percent in mice) [6][44], are essential players in innate immunity and inflammatory responses. Nearly every day, new neutrophils (1011 cells in human) are produced in the bone marrow, in a process called granulopoiesis [7][45]. Neutrophils were initially believed to be terminally differentiated, homogenous, and short-lived cells; however, recent studies challenged this hypothesis and showed that neutrophils have high diversity and plasticity, with regard to functions [8][9][10][11][12][13][14][15][16][17][46,47,48,49,50,51,52,53,54,55]. Notably, growing tumors strongly affect the development and activity of neutrophils via a plethora of released growth factors and cytokines [8][46].
Tumor-Associated Neutrophils
The role of neutrophils in cancer is still controversial. High numbers of immature myeloid cells in the blood, including neutrophils (neutrophilia), is a frequently seen phenomenon in cancer patients, as well as in tumor-bearing mice [8][18][19][46,56,57]. Neutrophils efficiently infiltrate to tumors (tumor-associated neutrophils, TANs) and constitute a very important component of the tumor microenvironment (TME) [20][21][58,59]. In numerous pre-clinical and clinical studies, TANs have been shown to participate in malignant transformation, anti-tumoral immunity, and angiogenesis [18][22][23][24][25][56,60,61,62,63]. Moreover, an increased neutrophil-to-lymphocyte ratio (NLR) is now a well-recognized predictive factor for the progression of many cancer types [26][27][28][29][30][64,65,66,67,68].
Strong body of evidence suggests that tumor cells produce growth factors (IL-3, GM-CSF, and G-CSF) or inflammatory cytokines (IL-1β, IL-6, IL-17 and TNF-α) to induce neutrophil production and support their survival [31][32][69,70]. Neutrophil-attracting chemokines, such as CXCL1, CXCL2, or CXCL8, are induced in hypoxic tumor tissue, through the activation of hypoxia-inducible factor 1 (HIF-1α or β) [8][19][33][46,57,71], and support neutrophil migration to the tumor site via CXCR1 and CXCR2 receptors [34][72]. Moreover, inflammatory cytokines stimulate G-CSF production in TME and further induce the generation of new neutrophils in bone marrow (cancer-related granulopoiesis) [35][36][73,74]. Neutrophils, in turn, contribute to cancer-related inflammation, thus promoting angiogenesis and tumor progression (Figure 1).
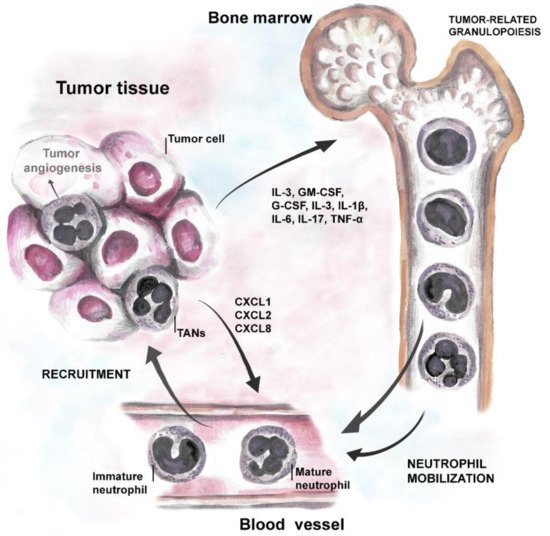
Figure 1. Tumor-neutrophil interactions. Tumor cells produce growth factors (IL-3, GM-CSF, and G-CSF) and inflammatory cytokines (IL-1β, IL-6, IL-17, and TNF) that induce neutrophil production and stimulate their survival. Chemoattractants specific for neutrophils, such as CXCL1, CXCL2 (ligands for CXCR2 on neutrophils), and CXCL8 (via CXCR1 on neutrophils) support their migration to the tumor site.
Tumor-associated neutrophils can exert ambivalent functions, ranging from direct killing of tumor cells to promoting tumor angiogenesis, metastasis, and immunosuppression. Similar to tumor-associated macrophages (TAMs), the phenotype and activity of TANs show significant heterogeneity throughout cancer development [37][38][39][40][41][75,76,77,78,79]. At the early stage of cancer, neutrophils are mainly located at the periphery of the tumor and display anti-tumoral phenotype, while, at later stages of the disease, their pro-tumoral properties dominate and support further cancer progression [19][42][43][44][45][46][47][57,80,81,82,83,84,85]. The pro- or anti-tumoral phenotype of neutrophils in the primary tumor or metastatic site is highly dependent on the cytokine milieu [48][86]. The functional polarization of TANs, under the influence of TME, and their phenotypic characterization have been previously reviewed in detail elsewhere [8][11][14][18][38][49][50][51][46,49,52,56,76,87,88,89]. It has been shown that IFN signaling triggers the anti-tumor activity of TANs (N1), characterized by normal density and hyper-segmented nucleus [17][52][55,90]. These cells express high levels of TNF-α, CCL3, and ICAM-1; additionally, they show low arginase-1 expression and are cytotoxic towards cancer cells in the experimental setting [53][91]. TGF-β stimulation or defective type I interferon (IFN) signaling stimulates the pro-tumoral activity of TANs (N2), with low density and both a segmented/not segmented nucleus [37][75]. These neutrophils can exert immunosuppressive activity, with upregulated expression of CCL2, CCL3, CCL4, CCL8, CCL12, CCL17, CXCL1, CXCL2, IL8/CXCL8, and CXCL16 chemokines [53][91]. N2 neutrophils promote the genetic instability of cancer cells by producing reactive oxygen species (ROS) and the release of exosomes containing microRNAs that impair nuclear integrity [8][46]. Moreover, upon G-CSF and TGF-β stimulation, neutrophils express arginase-1 (ARG1), ROS, and nitric oxide (NO) that contribute to the suppression of T cells [37][54][55][75,92,93]. Importantly, it has also been shown that both circulating, and tumor-associated neutrophils could suppress T cell activation by using immune checkpoint molecules, such as PDL-1 (programmed cell death ligand 1) or VISTA (V-domain immunoglobulin suppressor of T cell activation) [56][57][94,95].
3. Neutrophils and Tumor Angiogenesis
Neutrophils have been shown to modify their angiogenic capacity under the influence of tumor-derived factors, even before arriving at the TME [58][96]. Bone marrow neutrophils isolated from naive mice have been reported to suppress tumor growth, while their counterparts from tumor-bearing mice promoted tumor growth [58][96]. It is generally accepted that the pro-angiogenic properties of tumor-associated neutrophils support cancer progression. The involvement of TANs in tumor angiogenesis was already proposed, in 2006 by Nozawa et al., in a murine pancreatic islet carcinogenesis model. In this study, neutrophils were identified as an important source of MMP-9, and their functional involvement in tumor angiogenesis was demonstrated by the suppression of tumor angiogenic switch after depletion with the anti-Gr-1 antibody [59][97]. Following this study, the pro-angiogenic activities of neutrophils have been suggested in other cancer types, such as hepatocellular carcinoma, gastric cancer, melanoma, and fibrosarcoma [60][61][62][63][32,98,99,100]. In the liver tumorigenesis model, enhanced hypoxia and inflammation in TME have been shown to support the recruitment of neutrophils and promote tumor angiogenesis [61][98]. Nagaoka et al. demonstrated that IL-17-induced neutrophil infiltration, in a progressive tumor model of gastric cancer, increases tumor angiogenesis and supports tumor persistence [64][101]. Furthermore, the important role of neutrophil clustering in the vascularization of nasal cavity cancer (nasal inverted papilloma, NIP) was demonstrated recently [65][102]. MMP-9 and HIF-1α expressing neutrophils were shown to be the main tumor-infiltrating cells that promoted angiogenesis and significantly contributed to the NIP pathogenesis [65][102].
In agreement with the essential role of neutrophils in tumor angiogenesis, reduced tumor vascularization and growth have been demonstrated after antibody-mediated neutrophil depletion (using anti-Gr-1 or anti-Ly6G antibody) or inhibition of neutrophil infiltration into tumor tissue (e.g., anti-vascular cell adhesion molecule-1 and anti-VCAM-1) in several studies [60][59][63][32,97,100]. Adoptive transfer of anti-angiogenic neutrophils has also been shown to limit tumor angiogenesis in the murine model of melanoma [66][67][68][103,104,105], while the transfer of pro-angiogenic neutrophils supports tumor vascularization in mice [60][68][32,105].
Neutrophils have been recently shown to contribute to tumor vascularization, also through non-angiogenic mechanisms, vessel co-option, and vascular mimicry [69][70][106,107]. High neutrophil expression of LOXL4 in colorectal cancer liver metastases (mainly via vessel co-option) has been proposed to be the key factor responsible for the resistance against anti-angiogenic therapy [71][108]. In a very recent study, vascular mimicry (VM) structures (that consisted of cancer-associated fibroblasts (CAFs) and tumor cells) were shown to provide channels for neutrophil infiltration in lung cancer [70][107]. Such VM structures induced the pro-angiogenic (N2) polarization of infiltrating neutrophils and promoted their arginase, CCL2, CXCR4, and MMP-9 expression and, thus, evade anti-angiogenic therapy [70][107].
3.1. Pro-Angiogenic Switch of Neutrophils in Cancer
Neutrophils support the pro-angiogenic switch during cancer development [59][97] and are significant producers of pro-angiogenic factors in TME [72][73][109,110]. Pro-angiogenic factors that are released by neutrophils, such as VEGF, Bv8, MMP-9, and S100A8/S100A9, directly induce tumor angiogenesis via the activation of endothelial cell proliferation [60][12][59][74][32,50,97,111]. Human neutrophils have been shown to carry an intracellular pool of VEGF and mediate its rapid secretion, through the degranulation upon stimulation with phorbol-12-myristate 13-acetate (PMA) and TNFα [75][112] (Figure 2).
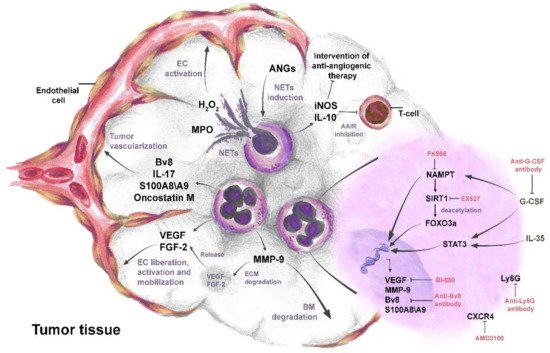
Figure 2. The complex role of neutrophils in tumor angiogenesis. IL-35- and G-CSF-mediated activation of STAT3 and NAMPT pathways leads to the up-regulation and secretion of pro-angiogenic factors VEGF, FGF-2, oncostatin M, IL-17, Bv8, MMP-9, and S100A8/S100A9 in neutrophils. The activated FOXO3a transcription factor also contributes to the production of pro-angiogenic factors. Pro-angiogenic factors released from neutrophils directly promote liberation, proliferation, and mobilization of endothelial cells (ECs) and induce tumor angiogenesis. Neutrophil MMP-9 mediates the degradation of the basal membrane (BM) and extracellular matrix (ECM). Degradation of ECM by MMP-9 leads to the release of sequestered VEGF and FGF-2 in ECM. Angiopoietins (ANGs) induce neutrophil extracellular trap (NET) formation in neutrophils. MPO in NETs increases H2O2 and stimulates proliferation and mobility of ECs. Neutrophils secrete IL-10 and iNOS to suppress anti-angiogenic adaptive immune response (AAIR) and later sustain tumor escape from anti-angiogenic therapy. Anti-Ly6G, anti-G-CSF, and anti-Bv8 antibodies inhibit neutrophil mediated tumor angiogenesis. Targeting NAMPT, SIRT1, VEGF, and CXCR4 in neutrophils, using molecular inhibitors such as FK866, EX527, BI-880, and AMD3100, respectively, reduces their pro-angiogenic activity.
In addition to their direct pro-angiogenic function, neutrophils can also activate pro-angiogenic functions of other immune cells and, thus, indirectly contribute to angiogenesis. It was previously demonstrated that human neutrophils can induce regulatory-like phenotypes of T cells and support their expression of IL-10, IL-17, and VEGF to promote vessel growth in pregnancy [76][113]. Possibly similar mechanisms could take place in the tumor microenvironment. Neutrophils could also affect anti-tumoral adaptive immune responses to support tumor angiogenesis [77][114]. In one study, Zou et al. identified pro-angiogenic immunosuppressive N2 neutrophils by producing iNOS inhibited T cell activation and suppressed anti-tumor adaptive immune responses, thus promoting tumor cell survival and proliferation [77][114].
Cancer metastasis constitute a major challenge in effective cancer therapy. Pro-angiogenic neutrophils have also been shown as significant supporters of tumor cell detachment and cancer metastasis [78][79][115,116]. In an animal model of breast cancer, JUNB deficiency was shown to increase neutrophil infiltration in the metastatic lung. Such neutrophils released elevated amounts of MMP-9 and BV8, leading to enhanced lung metastasis [80][117].
Importantly, neutrophils can also interfere with anti-angiogenic therapies. It was shown that the infiltration of CD11b+ Gr1+ myeloid cells (neutrophils) into tumor significantly reduced the efficacy of anti-VEGF antibody therapy [81][118]. Moreover, by secreting immunosuppressive cytokine IL-10 and hampering of T cell proliferation, neutrophils were shown to sustain tumor escape from anti-angiogenic therapy [82][119].
3.2. Neutrophil-Derived Factors That Support Tumor Angiogenesis
Neutrophils have been shown to support tumor angiogenesis via the release of numerous pro-angiogenic factors, such as VEGF, FGF-2, oncostatin M, IL-17, MMP-9, Bv8, S100A8/9 alarmins, STAT3, and NETs. Identification of other possible pro-angiogenic factors could support further approaches in successful cancer therapy.
3.2.1. VEGF and FGF-2
VEGF is a major pro-angiogenic factor, and VEGF signaling plays a central role in both physiological and pathological angiogenesis. It has been shown that human neutrophils carry an intracellular pool of VEGF and release it during the process of degranulation, upon stimulation with PMA, fMet-Leu-Phe (fMLP), and TNF-α [75][112]. Interestingly, neutrophils were found to be the main source of the circulating VEGF in the majority of patients with metastatic breast cancer or anal carcinoma [83][120]. Another study has reported that circulating neutrophils release higher amounts of VEGF in patients with oral cavity cancer, compared to healthy donor counterparts [84][121]. Furthermore, it has been shown that neutrophil depletion is linked to the inhibition of angiogenesis because of the reduced release of pre-formed neutrophil VEGF stores [85][122]. Importantly, elevated infiltration of tumor tissue by CD11b+Gr+ neutrophils, characterized by the increased expression of VEGF, was demonstrated to contribute to tumor angiogenesis in a mouse model of fibrosarcoma and melanoma [60][32].
Surprisingly, VEGF was also shown to indirectly affect neutrophil recruitment. It was reported that IL-8 expression by endothelial cells after VEGF stimulation induced neutrophil migration in vitro [86][123]. Moreover, VEGF production by neutrophils might promote the further recruitment of neutrophils, in a positive feedback loop, to establish pro-angiogenic signaling inside the tumor and support tumor angiogenesis.
Additionally, neutrophils have been shown to produce and/or contribute to the release of FGF-2. The direct FGF-2 expression of tumor-infiltrating neutrophils was demonstrated in the mouse model of hepatic metastases from gastrointestinal tumors [79][116]. In this study, neutrophils from hepatic metastases were shown to abundantly express FGF-2 mRNA and promote angiogenesis. Furthermore, neutrophil activity stimulates the release and bioavailability of sequestered FGF-2 in the extracellular matrix [87][88][124,125].
3.2.2. Oncostatin M and IL-17
Oncostatin M is a cytokine produced by T cells, monocytes, and neutrophils. In early studies, oncostatin M has been shown to inhibit cancer cell proliferation and tumor growth [89][90][126,127]. However, neutrophil-secreted oncostatin M has been suggested to contribute to angiogenesis and invasiveness of breast cancer [78][115]. When co-cultured with breast cancer cells in vitro, human blood neutrophils have been shown to express oncostatin M [78][115]. Furthermore, both the cell–cell contact and GM-CSF production of breast cancer cells were demonstrated to be necessary for the oncostatin M release from neutrophils. Importantly, GM-CSF-dependent production of oncostatin M by neutrophils has been shown to promote the invasiveness and VEGF expression of MDA-MB-231 and T47D human breast cancer cells [78][115].
Neutrophil-derived IL-17 has also been suggested to induce tumor angiogenesis [91][128]. Numasaki et al. demonstrated that IL-17 enhances the angiogenic activity and in vivo growth of non-small cell lung cancer (NSCLC) by promoting the expression of angiogenic chemokines (CXCL1, CXCL5, CXCL6, and CXCL8) [91][128]. Interestingly, tumor infiltrating T cells and neutrophils were identified as the major source of IL-17 in NSCLC tissue [91][128].
3.2.3. MMP-9
MMPs are reported to be highly expressed in various cancer types and are commonly associated with the tumor progression [92][93][94][129,130,131]. MMP-9 is a gelatinase that mediates the remodeling of the extracellular membrane, and it has been demonstrated to play an important role in tumor angiogenesis by triggering the angiogenic switch [95][132]. Importantly, tumor-associated neutrophils were reported to have significantly more abundant MMP-9, readily available for rapid release, than tumor-associated macrophages [96][133]. Neutrophil-produced MMP-9 mediates degradation of the basal membrane and leads to the liberation and activation of ECs. The degradation of the basal membrane, by MMP-9, indirectly leads to the release of pro-angiogenic factors VEGF and FGF-2, stored in extracellular matrix [59][79][88][97][97,116,125,134]. VEGF, sequestered in the tumor extracellular matrix, after being released by MMP-9, increases further neutrophilic expression of MMP-9 in a positive feedback fashion [98][135]. VEGF-A was also demonstrated as a chemoattractant for MMP-9-expressing neutrophils [99][136]. Moreover, increased levels of VEGF-A in hypoxic regions recruit a distinct subpopulation of pro-angiogenic neutrophils that are characterized by the expression profile of CD49d+VEGFR1hiCXCR4hi [100][137]. Neutrophil MMP-9 also increases VEGF-A bioavailability and bioactivity and contributes to the inflammation-induced lymphangiogenesis [101][138]. Li et al. recently identified high levels of IL-17+ neutrophils as the major source of MMP-9 in the invasive margin of gastric cancer [102][139]. In this study, IL-17 producing neutrophils were shown to promote the pro-angiogenic activity of cancer cells, both in vivo and in vitro. Moreover, such neutrophils stimulated cancer cells to produce neutrophil chemoattractants, which, in turn, supported further neutrophil recruitment [102][139]. The pro-angiogenic role of MMP-9-secreting neutrophils in tumor vascularization was also validated in other types of cancer. In primary tumors formed by highly disseminating variants of human fibrosarcoma and prostate carcinoma, an elevated accumulation of MMP-9-positive neutrophils was demonstrated. The inhibition of neutrophil recruitment into such tumors, by IL-8 neutralization, diminished tumor angiogenesis [103][140].
3.2.4. Bv8 and S100A8/9 Alarmins
Neutrophil-derived Bv8 and S100 proteins (S100A8 and S100A9) increase tumor proliferation and angiogenesis by supporting neutrophil mobilization, EC activation, and proliferation [8][104][105][106][46,141,142,143]. Tumor-associated neutrophils have been shown to produce Bv8 and S100A8 in a mouse melanoma model [68][105]. Bv8 has been suggested as a modulator of myeloid-cell-dependent tumor angiogenesis, and neutrophils have been reported to produce Bv8 robustly, after stimulation with G-CSF or GM-CSF [105][107][142,144]. Recently, Itatani et al. demonstrated that tumor-infiltrating neutrophils strongly expressed Bv8 in a mouse model of colorectal cancer. Importantly, in the same study, significantly higher plasma levels of Bv8 were observed in CRC patients, compared to healthy donors. Moreover, high plasma levels of Bv8 positively correlated with a bad prognosis and decreased overall survival of such patients [108][145]. In another study, treatment with anti-Bv8 antibodies reduced the number of angiogenic islets in transgenic mouse model of multistage pancreatic β-cell tumorigenesis [74][111]. Moreover, such anti-Bv8 treatment inhibited the recruitment of CD11b+Gr+ neutrophils to the neoplastic lesions [74][81][109][111,118,146]. Apart from direct stimulation of tumor angiogenesis, neutrophil-derived Bv8 also plays a role in tumor resistance to anti-VEGF therapy (see chapter 7) [108][145]. Increased levels of plasma G-CSF, after anti-VEGF therapy of mice bearing VEGF-resistant colorectal tumors, were shown to promote neutrophil infiltration into tumor stroma and stimulate angiogenesis via elevated neutrophil Bv8 expression [108][145]. This study demonstrates that blocking neutrophil-derived Bv8 increases the efficacy of anti-VEGF antibody therapy in colorectal cancer [108][145].
3.2.5. STAT3
STAT3 is transcription factor that is involved in cell proliferation, survival, and angiogenesis [110][147]. STAT3 has been reported to be constitutively activated in both tumor and immune cells in the tumor microenvironment [110][111][112][147,148,149]. Neutrophil STAT3 activity (triggered via G-CSF/G-CSFR pathway) is important for their production, survival, and mobilization from the bone marrow [113][114][115][150,151,152]. STAT3 signaling in mice has been shown to have a regulatory role in neutrophil pro-angiogenic activity by controlling the expression of VEGFA, FGF-2, and MMP-9 genes [116][153]. Importantly, it has also been demonstrated that neutrophils isolated from tumors have activated STAT3 and induce angiogenesis in vitro [116][153]. STAT3 signaling in neutrophils was suggested to activate neutrophil pro-angiogenic functions, even before they entered the TME [58][96]. Stimulation by G-CSF and IL-6 was shown to increase Stat3 expression, which then led to the upregulation of MMP-9 and Bv8 genes in bone marrow neutrophils [58][96]. Mechanistically, G-CSF-mediated STAT3 activation induces binding of phospho-STAT3 to the Bv8 promotor and elevates the expression of Bv8 [117][154]. The inhibition of Stat3, by type I IFN signaling, impairs VEGF and MMP-9 production by neutrophils and suppresses tumor angiogenesis [60][32].
3.3. The Role of Neutrophil Extracellular Traps in Angiogenesis
Neutrophils contribute to inflammatory angiogenesis, also by neutrophil extracellular trap (NET) formation. Myeloperoxidase that is attached to such NETs increases H2O2 in ECM and leads to the activation of NFκB-mediated inflammatory signaling in ECs via TLR4. This initiates proliferation and mobility of endothelial cells and, as a result, tumor angiogenesis [118][155]. It has previously been shown that cancer cells are able to induce NET formation by neutrophils and, thus, to promote tumor angiogenesis [119][156]. Angiopoietins (ANG-1 and ANG-2) also were shown to induce NETs formation and contribute to pro-inflammatory and pro-angiogenic activities of neutrophils [120][157]. Angiopoietin-induced NETosis of human neutrophils promoted neutrophil adhesion to HUVECs and stimulated their proliferation [120][157].