Non-alcoholic fatty liver disease (NAFLD) is a slowly progressing disease, beginning with isolated liver steatosis that evolves in a subset of patients to non-alcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC). It was recently proposed to redefine NAFLD to metabolic dysfunction-associated fatty liver disease (MAFLD) in which other known causes of liver disease such as alcohol consumption or viral hepatitis do not need to be excluded. Revised nomenclature envisions speeding up and facilitating anti-MAFLD drug development by means of patient stratification whereby each subgroup would benefit from distinct pharmacological interventions.
1. Introduction
The prevalence of non-alcoholic fatty liver disease (NAFLD) has risen over past decades, closely paralleling the obesity pandemic, making it the number one chronic liver disease today [
1]. NAFLD is a slowly progressing disease, beginning with isolated liver steatosis that evolves in a subset of patients to non-alcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC) [
2]. NAFLD is defined as the presence of hepatic steatosis in at least 5% of the liver parenchyma, in the absence of other hepatic pathologies, such as viral hepatitis and without significant alcohol consumption or intake of steatogenic drugs (e.g., valproic acid, tamoxifen) [
3].
Major risk factors for developing NAFLD are obesity and type II diabetes mellitus (T2DM), which is why NAFLD is often considered as the hepatic manifestation of the metabolic syndrome. NAFLD patients typically exhibit serum lipid dysregulation characterized by increased levels of low-density lipoprotein (LDL) particles and a reduced amount of high-density lipoproteins (HDL) [
4,
5]. Unsurprisingly, cardiovascular disease is one of the most common causes of death in NAFLD patients. Because of the unmet need for effective anti-NAFLD therapeutics, hundreds of clinical studies have been carried out in the past decade, however, without satisfying results [
6]. Clinical studies report drug-response rates ranging from 20–40% with placebo effects accounting for 10 to 20% [
7,
8], suggesting the presence of certain effect modifiers.
Weight loss through lifestyle modification, achieved through close follow-up by dieticians and physical activity, might be a cost-effective and efficient approach for treatment of NAFLD [
9]. Consequently, one solution to avoid noise in clinical studies could be standardization of diet and exercise during anti-NAFLD treatment [
10]. Yet, one may not forget that certain NAFLD/NASH patients are lean or exhibit other non-obesity related causes of NAFLD for which weight loss is not an option. Further, the desired lifestyle modifications and weight loss are often hardly attainable outside well-controlled settings. In addition, it has become clear that NAFLD should be better understood and that its pathogenesis is likely much more heterogenic than previously assumed [
11]. In this context, the NAFLD nomenclature was recently revised to ‘metabolic dysfunction-associated fatty liver disease (MAFLD)’, in order to avoid diagnosis based on exclusion of other pathologies and to better mirror its pathogenesis [
7].
The diagnosis of MAFLD is made based on the presence of hepatic steatosis with at least one of three of the following features: (i) obesity/overweight, (ii) diabetes mellitus and/or (iii) evidence of metabolic dysregulation [
7,
12]. The origin of MAFLD is, however, unspecified and highly heterogeneous due to multiple coexisting disease-modifiers including sex, age, diet, alcohol consumption, concurring liver pathology, genetic background and metabolic pressure. Most patients suffering from NAFLD also meet the criteria for MAFLD. A recent cross-sectional study revealed that the NAFLD prevalence was 37.1% (95% CI, 34.0–40.4), whereas the MAFLD prevalence was slightly higher at 39.1% (95% CI, 36.3–42.1) in the general population of the United States of America [
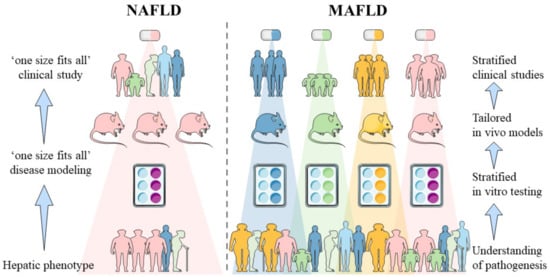
13]. Therefore, patient heterogeneity should also be adopted in preclinical disease modeling and drug screening studies. The impact of the change in terminology on different stages of drug development is visualized in
Figure 1.
Figure 1. Impact of MAFLD nomenclature on different stages of drug development. Until now, patient inclusion for anti-NAFLD clinical studies was based on hepatic phenotype in the absence of significant alcohol intake and concurring pathologies. Anti-MAFLD trials will be undertaken based on pathogenic determinants that will likely require distinct pharmacological interventions. Introduction of the MAFLD terminology requires deeper understanding of the pathogenesis of fatty liver disease and hence necessitates the development of human-based in vitro models able to represent the distinct disease subsets. Disease subsets of MAFLD are indicated by the blue, green, yellow and pink colors. Corresponding in vitro tests, in vivo models, stratification in clinical studies and distinct pharmacological interventions are visualized by the same color for each disease subset. [Abbreviations: MAFLD: Metabolic-dysfunction associated fatty liver disease, NAFLD: non-alcoholic fatty liver disease].
2. Sources of Heterogeneity in MAFLD and Their In Vitro Implementation
2.1. Sex and Hormonal Status
NAFLD occurs more often in males, in whom higher liver pyruvate kinase levels positively correlate with NAFLD severity [
33,
34]. A recent transcriptomic study also points to the fact that NASH is a sexual dimorphic disorder that might lead to different drug responses between men and women [
35]. Remarkably, the phase II trial in which the dual C-C motif chemokine receptor (CCR) 2/5 antagonist cenicriviroc was evaluated showed a positive effect in men regarding fibrosis regression after one year, but not in women, although this was not necessarily associated with response to cenicriviroc [
36]. Based on these findings, it can be presumed that anti-NASH treatment likely requires a sex-specific approach and thus the need for both female- and male-based preclinical models. Moreover, as gender-affirming hormone therapy has also been shown to lead to altered metabolic profiles [
37], but little is known about the impact of such therapies on MAFLD development, in vitro models might prove useful to gain further insights on this topic. As sex hormones such as estrogens and androgens are known to influence lipid metabolism [
38], their inclusion in in vitro MAFLD models could be a first step towards sex-specific MAFLD models.
2.2. Pediatric/Juvenile MAFLD
NAFLD prevalence positively correlates with aging [
34,
41,
42]; however, pediatric NAFLD seems a distinct entity with a more severe phenotype [
41]. Indeed, NASH-related cirrhosis has been reported already in children of eight years old [
42]. Pediatric NAFLD patients exhibit greater periportal inflammation, which is associated with hyperuricemia, while adult NAFLD mainly manifests in perivenous zones [
43]. Pediatric periportal steatosis associates with advanced fibrosis, while perivenous steatosis correlates with steatohepatitis. Of these subgroups, children suffering from periportal steatosis are significantly younger than the ones suffering from perivenous steatosis (ten vs. fourteen years old, respectively) [
44].
Reportedly, 8% of children from the general population and up to 34% of obese children suffer from NAFLD globally [
45,
46,
47]. Between 1990 and 2017 the annual increase in juvenile NAFLD prevalence was 1.35% and although this increasing trend is observed worldwide, it is most pronounced in regions with a high-middle socio-demographic index [
47]. Considering the increasing prevalence of NAFLD among children, in vitro models could provide insights in the underlying mechanisms that drive pediatric MAFLD and aid drug development for this specific subgroup. One way to achieve this is to use iPSC generated from cells collected from children in a non-invasive way (e.g.
, urine cells) [
48,
49].
2.3. Fructose Consumption
In the past decade, excessive dietary fructose consumption has been identified as a driver of NAFLD development and progression. Fructose-related liver steatosis and NASH are independent from metabolic syndrome or obesity, which makes it a silent and widely spread risk factor [
50,
51,
52]. Fructose intake is, however, associated with the development of features of the metabolic syndrome, such as elevated serum triglycerides and insulin resistance [
53,
54]. Fructose is metabolized in vivo by the liver and therefore it can be easily introduced to current in vitro models by direct (sequential) exposure of the hepatic cells through the culture medium [
55]. Indeed, human co-cultures consisting of primary hepatocytes, stellate cells, hepatic sinusoidal endothelial cells and biliary epithelial cells have been exposed to glucose and fructose and shown to exhibit increased expression of lipogenic and fibrogenic genes. This can be reversed by inhibiting ketohexokinase, which intervenes in the enzymatic phosphorylation of fructose to fructose-1-phosphate [
56]. Nonetheless, not all NASH-promoting effects of fructose are mediated by the aforementioned cell types. Fructose also promotes endotoxemia and activation of MYD88 innate immune signal transduction adaptor (MYD88)-mediated inflammatory signaling in liver myeloid cells resulting in the generation of tumor necrosis factor alpha (TNF-α). TNF-α subsequently stimulates caspase 2 and endoplasmic reticulum (ER)-stress, which in turn triggers the lipogenic transcription factor sterol regulatory element-binding protein-1c (SREBP-1c) to initiate de novo lipogenesis. Thus, modeling mechanisms of fructose-mediated MAFLD requires more complex disease models that include different cell types, or well-defined culture media that mimic the presence of non-parenchymal cells, likely consisting of fructose, endotoxins and TNF-α, of which the latter seems the most important trigger of lipogenesis [
17].
2.4. Genetic Predisposition and Ethnicity
Important racial and ethnic disparities exist in NAFLD prevalence and risk for progression to end-stage liver disease [
57]. The prevalence of NAFLD is the highest among the Hispanic population, the lowest in the black population and intermediate in the white population. Similarly, in comparison to white people, the risk for progression to NASH is the highest in Hispanic people (relative risk, 1.09; 95% confidence interval (CI), 0.98–1.21) and the lowest in black individuals (relative risk, 0.72; 95% CI, 0.60–0.87) in comparison to the white population, although without a difference in fibrosis occurrence [
58]. The carriage of certain single nucleotide polymorphisms (SNPs) has also been linked to NAFLD development and disease severity. Hitherto, the carriage of
patatin-like phospholipase domain-containing protein 3 (
PNPLA3) rs738409 appears to be the strongest genetic disease modifier [
59]. Other SNPs that have been linked to NASH development and progression, include
transmembrane 6 superfamily member 2 (
TM6SF2) rs58542926, involved in very low-density lipoprotein (VLDL) lipidation and secretion, and
membrane bound O-acyltransferase domain containing 7 (
MBOAT7) rs641738, involved in phospholipid acyl-chain remodeling [
60,
61,
62,
63]. Of note,
PNPLA3 rs738409 is highly prevalent among the Hispanic population [
58]. Previous in vitro and clinical research showed that PNPLA3 rs738409 has desaturating activity towards liver triglycerides, because it impairs hydrolysis/transacylation of poly-unsaturated fatty acids from diacylglycerols for synthesis of phosphatidylcholine [
64]. In addition, homozygous carriage of
PNPLA3 rs738409 promotes the pro-fibrogenic properties of hepatic stellate cells [
65]. Therefore, cellular systems that carry this specific polymorphism are of particular interest to model MAFLD and could enable personalized testing of anti-NASH pharmaceuticals. Recent in vitro research, using induced pluripotent stem cell (iPSC)-derived hepatocyte-like cells with knocked in
PNPLA3 rs738409, showed that this polymorphism induces a loss of function of PNPLA3 and thus impairs lipid metabolism, thereby predisposing these cells to steatosis. Furthermore, due to downregulation of enzymes involved in xenobiotic metabolism and excretion,
PNPLA3 rs738409 also increases sensitivity to hepatotoxins. This could explain how subclinical but sustained hepatic damage, attributed to environmental hepatotoxins, may result in disease progression towards NASH in patients carrying
PNPLA3 rs738409 [
66]. For these reasons, cellular models for either dissecting the MAFLD pathogenesis or for testing novel therapeutics should be genetically characterized. As the ultimate example of in vitro translation of patient heterogeneity, it was demonstrated that iPSC-derived hepatocyte-like cells from four different donors, overloaded with oleic acid, also showed different lipid metabolism associated gene expression profiles related to the steatosis phenotype of the donor [
20].
2.5. Epigenetics
Epigenetic alterations of gene expression, including DNA methylation, histone modifications and non-coding microRNA (miR) expression, are involved in underlying disease mechanisms of NASH and other metabolic diseases [
67]. Liver miR-122 expression is 10-fold lower in NASH patients in comparison to simple steatosis, while in vitro its overexpression in transfected Huh7 cells results in higher alanine aminotransferase activity [
68]. Various other miRs have also been associated with NASH, such as miR-223, which is elevated in NASH liver biopsy samples [
69].
NASH patients exhibit significantly elevated levels of liver mitochondrial DNA methylation [
70] and the latter is also involved in transdifferentiation of hepatic stellate cells to myofibroblasts, hence accompanying liver fibrosis [
71]. Moreover, DNA methylation profiles could serve as a prognostic tool for NAFLD in the prevention of disease progression towards HCC [
72].
Tubulin, beta 2B class IIB (
Tubb2b) was found to be hypomethylated and overexpressed in a murine model investigating NASH-associated liver carcinogenesis. Markedly,
Tubb2b was increasingly demethylated in the stages of NAFLD, NASH-fibrosis and HCC and this inversely correlated with
Tubb2b gene expression at all of these stages. This phenomenon was also more present in HepG2 cells when compared to HepaRG cells, the latter being considered as better-differentiated hepatic cells. When culturing HepaRG cells in the presence of oleic acid, methylation of the
Tubb2b promotor decreased along with increased expression of the gene. Overall, these data suggest that
Tubb2b overexpression, due to increased demethylation, may be involved in the NAFLD pathogenesis and development of HCC [
73].
2.6. Obesity and Body Fat Distribution
Obesity is one of the most common and well described risk factors for NAFLD [
77]. Indeed, among adult NAFLD patients, the prevalence of obesity is estimated at 51.34% (95% CI, 41.3–61.20%) and the global increase in NAFLD prevalence is consistent with the alarming rise of obesity prevalence worldwide [
78]. However, a subgroup of obese patients can be categorized as metabolically healthy and exhibit a significantly decreased risk of developing comorbidities [
79,
80]. Contrarily, up to 30% of adult normal weight individuals are metabolically obese and display increased cardiometabolic risk [
81,
82]. Therefore, it seems it is not the total amount of body fat but rather the distribution thereof that is an important disease driver. Indeed, visceral adipose tissue (VAT) area is associated with increased risk to develop NAFLD. To the contrary, larger areas of subcutaneous adipose tissue were found to associate with NAFLD regression in normal weight individuals [
83]. Further, VAT area directly relates to hepatic inflammation and fibrosis development in NAFLD patients [
84,
85]. VAT is a hormonally active tissue that releases a spectrum of mediators that are involved in NAFLD such as adiponectin, interleukin 6 (IL-6) and TNF-α. Serum level of adiponectin, an anti-inflammatory and anti-lipogenic adipokine, is negatively correlated with VAT area [
86] and was also found to be decreased in NASH patients [
87]. In patients with metabolically unhealthy obesity, increased concentrations of IL-6 were found in VAT, suggesting increased pro-inflammatory activity of VAT [
88,
89]. Serum concentrations of IL-6 were also determined to significantly relate to VAT area [
90]. Furthermore, IL-6 content in adipose tissue of metabolically unhealthy obese patients was found to be over one hundred-fold higher when compared to the liver content, suggesting that adipose tissue is the key source of IL-6 during metabolic dysregulation [
88]. The degree of hepatitis and fibrosis positively correlates with both circulating and liver IL-6 concentrations [
91]. From these data, it is clear that adipose tissue, and in particular VAT, is an important disease driver and should be encompassed in MAFLD in vitro
models. A basic approach to recapitulate the influence of VAT-secreted mediators would be to expose human cell cultures to physiologically relevant concentrations of adiponectin, IL-6 and TNF-α in vitro. A recently proposed microfluidic device consisting of primary human hepatocytes and adipocytes was able to demonstrate indirect effects of adipose tissue on hepatocytes and seems promising for use in preclinical settings [
92].
2.7. Lean MAFLD
Approximately 30% of normal weight individuals are metabolically unhealthy, which is also known as metabolic obesity. These persons exhibit a higher risk to develop metabolic diseases such as MAFLD [
81,
82]. In lean individuals worldwide, the NAFLD prevalence is 9.7% (95% CI: 7.7–11.8%), which is, however, in absolute proportions a significant number of NAFLD patients. Middle-aged patients in Asian countries show the highest prevalence of lean NAFLD [
93]. Lean NAFLD patients often suffer from metabolic dysfunction and visceral adiposity [
94], but also exhibit altered gut microbiota, higher secondary bile acid and FGF19 levels along with lower 7-alpha-hydroxy-4-cholesten-3-one compared to healthy persons and patients suffering from non-lean NAFLD [
95]. Interestingly, when compared to obese NAFLD patients, the prevalence of the SNP
TM6SF2 rs58542926 seems to be higher in individuals suffering from lean NAFLD [
95,
96]. From these data, it is clear that lean MAFLD should be considered as a distinct entity with more than one metabolic determinant. Importantly, lean NAFLD in white individuals may progress to end-stage liver disease without associated
PNPLA3 rs738409 variant or progression to obesity, along with the full spectrum of metabolic comorbidities [
97]. The true determinants of lean MAFLD are, however, still unknown. Mechanistic and translational in vitro studies regarding genetic determinants and drug response in this patient group therefore seem of high importance.
2.8. Microbiota
In the past decade, the gut-liver axis gained much attention as a key player in a variety of diseases.
Proteobacteria, Enterobacteria, Escherichia [
98] and
Bacteroides [
99] have been found to be more abundant in the stool of adult NASH patients than in healthy individuals. In stool samples of children with NAFLD,
Gammaproteobacteria were found to be increased when compared to both obese and non-obese children without NAFLD [
100]. Of note, NASH patients were found to exhibit increased serum levels of endogenous ethanol. Indeed, alcohol-producing microbiota such as
Escherichia are more abundant in the stool of NASH patients [
98]. Another study found that in a cohort of Chinese NAFLD patients, up to 60% of the patients were associated with high-alcohol-producing
Klebsiella pneumoniae [
101]. These findings suggest a role for these microbiota in the pathogenesis of NAFLD. Translocation of bacterial components to the liver can initiate and push MAFLD development. The best-known factor implicated in gut-related NASH is lipopolysaccharide (LPS) derived from Gram-negative bacteria [
102]. LPS can induce hepatic inflammation by interacting with Toll-like receptors (TLRs) on both hepatocytes and hepatic stellate cells. For example, primary human hepatocytes challenged with LPS in vitro exhibited upregulation of IL-1β and IL-6 [
103]. Additionally, upon exposure to LPS in vitro
, LX-2 stellate cells displayed upregulation of
IL-1β,
IL-6,
TGF-β and
TNF-α, highlighting the proinflammatory effects of LPS [
104]. Although in vitro direct exposure of cell systems to LPS has been demonstrated to be a practical approach [
105], a more comprehensive way of working could be investigated. Indeed, one of the possibilities to more accurately model dysbiosis-driven MAFLD could be to expose human cell cultures to bacterial metabolites that are found at higher levels in the plasma of MAFLD patients, such as trimethylamine N-oxide, glycocholic acid and deoxycholic acid [
106].
2.9. Zonation
The liver exhibits metabolic zonation [
107]. Hepatocytes located close to the portal triad (zone 1) are more involved in gluconeogenesis and fatty acid β-oxidation, while hepatocytes in the pericentral region (zone 3) are rather involved in glycolysis and lipogenesis [
108]. As such, steatosis generally occurs in the pericentral regions where the oxygen concentration is relatively lower [
109]. However, in certain pediatric NAFLD patients, steatosis occurs in the periportal region and can be azonal as well [
44]. Although lipid zonation seems lost in NASH [
110], future research should clarify whether certain MAFLD phenotypes are of ‘pediatric’ or ‘adult’ nature and how oxygen tension and nutrient supply could modify lipogenesis in certain cell types. Furthermore, cells are commonly cultivated under 20–21% oxygen, while the partial oxygen tension in the liver ranges from 10–12% (periportal) to 3–5% (pericentral) [
111]. This raises the question whether standard cultivation techniques for hepatic cells, whether or not for MAFLD research, should be revised [
112]. As microfluidic devices support inclusion of zonation by limiting the flow rate, hereby altering oxygen and nutrient supply, they could provide valuable insights in this area of MAFLD research.
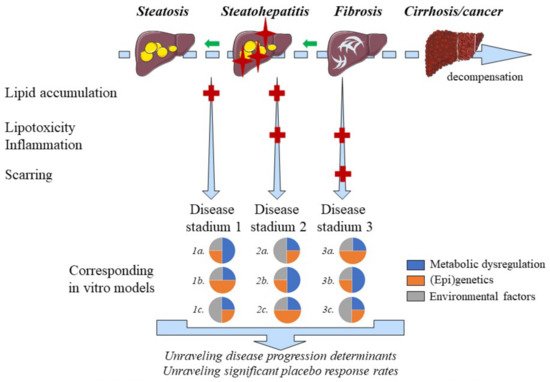
2.110. Disease Progression and Regression
MAFLD is a naturally progressing and regressing disease, which may partly explain high placebo response rates in clinical studies. In vitro models are well-positioned to study the MAFLD disease course, which seems key for unraveling the determinants of disease progression, regression and hence high placebo response rates (
Figure 2) [
2]. Hepatic steatosis is considered to precede inflammation across the spectrum of MAFLD. Yet, inflammation may also precede lipid accumulation [
88], which is in conflict with traditional presumptions. Using iPSC-derived hepatic cells it was demonstrated that thapsigargin-mediated induction of ER-stress is an important driver of lipogenesis [
113] and a pathway that should be considered when modeling steatosis-preceding inflammation. Stressed hepatocytes may accumulate lipids, which can easily be modelled in vitro using co-culture systems with macrophages and/or stellate cells [
16]. Yet, increasing complexity, which is typical in co-cultures, goes along with decreased large-scale applicability.
Figure 2. Positioning of MAFLD disease models. MAFLD is a naturally progressing and regressing disease of which different stadia can be accurately modeled in vitro. The main pathogenic drivers, however, differ among patients suffering from MAFLD, which requires multiple disease models for different MAFLD stadia. Steatosis is indicated by yellow ovals whereas red crosses on the liver figures indicate additional inflammation. Light blue fibers indicate hepatic accumulation of extracellular matrix components. The key factors involved in the different stages of MAFLD, mentioned on the left side of the figure, are indicated by corresponding red ‘+’ symbols. Regression of the disease is represented by the green arrows in the decompensation timeline.
2.121. Ethanol consumption
Conflicting data exists regarding the effects of ethanol consumption in patients suffering from NAFLD. A prospective cohort study including 4568 NAFLD patients concluded that modest ethanol consumption (i.e., 0.5 to 1.5 standard drinks per day) decreases all-cause mortality compared to ethanol abstinence. In contrast, drinking more than 1.5 standard drinks of ethanol per day, rapidly increases all-cause mortality [125]. These findings are consistent with the fact that moderate ethanol intake reduces the risk for several cardiovascular outcomes [126] and that the beneficial or detrimental effects of ethanol on different health aspects follow a ‘J-shaped’ curve [125]. Prohibiting ethanol consumption in clinical studies is far from practical, often does not reflect the daily situation and would narrow the definition of MAFLD [7,127]. Therefore, pharmacological effects of novel anti-NASH compounds could be evaluated in vitro with different alcohol concentrations to gain insights in situations that reflect daily life for many patients. Ethanol induces transcriptional activity of SREBP-1c and carbohydrate-responsive element-binding protein thereby promoting lipogenesis [128]. SREBP-1c levels, however, decrease in patients with NASH progression [129]. Concomitant exposure of FaO hepatoma cells to ethanol and free fatty acids reduces lipid droplet size compared to single treatments [130], which correlates with more advanced MAFLD histology [131]. Coexisting alcoholic- and non-alcoholic fatty liver disease might therefore be an important but underrecognized disease. Evaluation of compounds using in vitro systems, in which both the effects of ethanol and metabolic factors are modeled, can facilitate the development of drugs targeting mixed pathologies.