Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Beatrix Zheng and Version 2 by Beatrix Zheng.
Tolvaptan-treated ADPKD patients have reduced OxSt levels compared to untreated patients. This effect may contribute to the slowing of renal function loss observed with tolvaptan treatment.
- ADPKD
- oxidative stress
- tolvaptan
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic multisystem disorder with an autosomal dominant pattern and an estimated prevalence of affected subjects between 1:400 and 1:1000. It is characterized by multiple, bilateral renal cysts and is found in 10% of patients with end-stage renal disease (ESRD).[1]
ADPKD is caused by mutations in the PKD1 (78% of cases) or PKD2 (15% of cases) genes. PKD1, located on chromosome 16 (16p13.3), encodes polycystin-1 (PC1), a large multidomain glycoprotein that is cleaved at a proteolytic site of the G protein-coupled-receptor. PKD2, located on chromosome 4 (4q21), encodes for polycystin-2 (PC2), a protein belonging to the transient receptor potential family of calcium-regulated cation channels.
Both PC1 and PC2 are found on primary cilia and studies now support an inhibitory role of PC1 and PC2 against cystogenesis [2]. Loss of PC1 or PC2 is associated with low intracellular calcium concentrations, which lead to increased activity of adenyl cyclase type 5 and 6, reduced activity of phosphodiesterase 1, excessive concentrations of cyclic AMP (cAMP), and consequent cystogenesis through the activation of the proliferation and secretion pathways [2].
Preclinical studies showed the role of arginine-vasopressin-mediated cAMP signaling as a driver of cystic proliferation and fluid secretion in ADPKD. In mouse models, the suppression of vasopressin release, vasopressin V2 receptor antagonism, or genetic elimination of vasopressin resulted in improved cyst burden [3].
Tolvaptan, a vasopressin V2 receptor antagonist, was shown to be effective in slowing the increase in total renal volume during a 3-year study period and it is now a consolidated treatment option for ADPKD patients [4].
Oxidative stress (OxSt), inflammation and endothelial dysfunction are non-traditional cardiovascular risk factors that have a key role in the induction of atherogenesis, malnutrition, and anemia in patients with chronic kidney disease (CKD). OxSt is one of the main factors leading to the progression of CKD [5] [6]. Increased OxSt in patients with CKD has been linked to a reduction in antioxidants and reduced bioavailability of nitric oxide (NO) [7][8][9]. The RhoA/Rho kinase (ROCK) system is deeply involved in a broad spectrum of essential physiological processes, including having a key role in the induction of OxSt signaling [5][10][11]; the activation of the RhoA/Rho kinase pathway essentially leads to cardiovascular-renal remodeling via the induction of OxSt and the reduction in NO bioavailability [5][10][11][12].
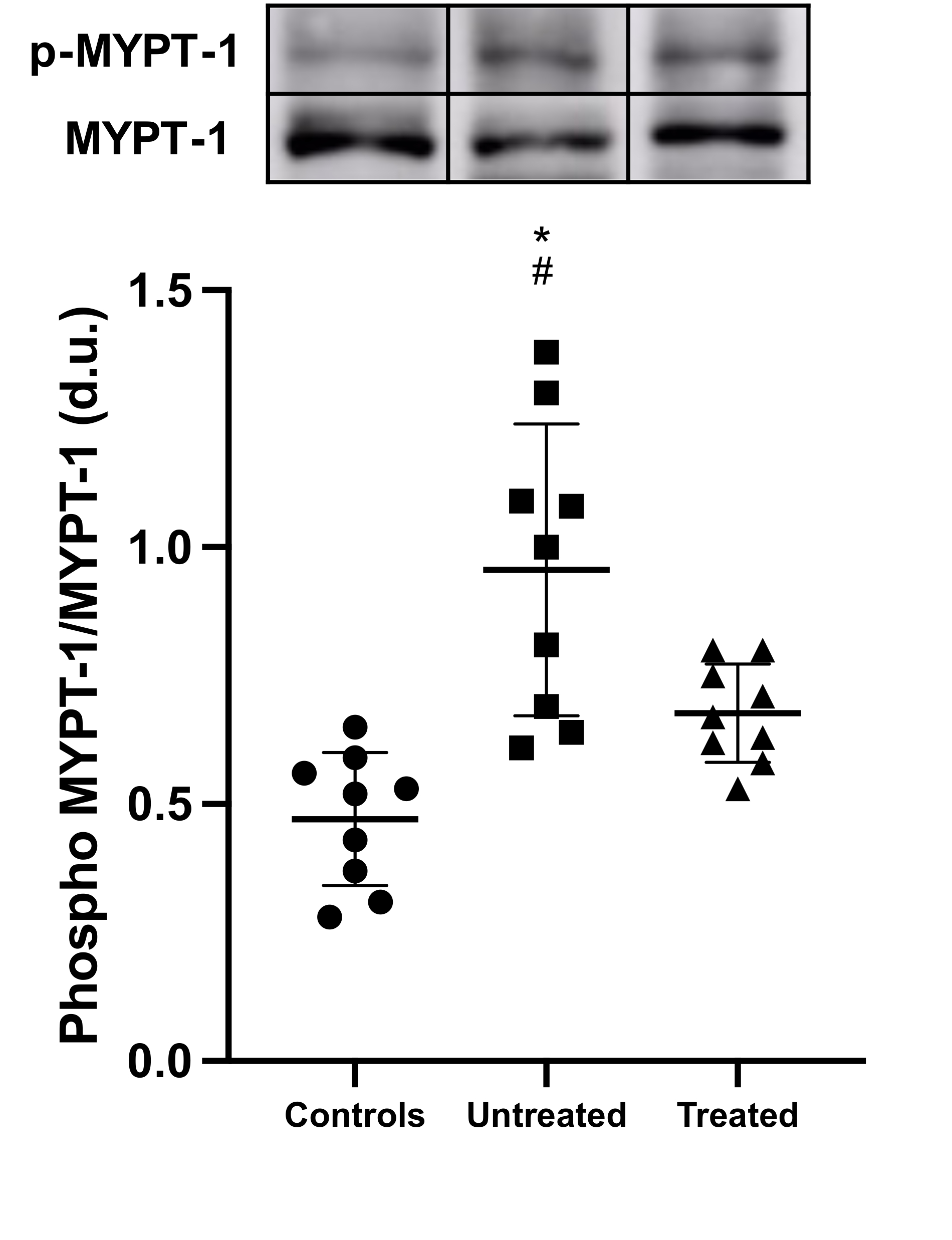
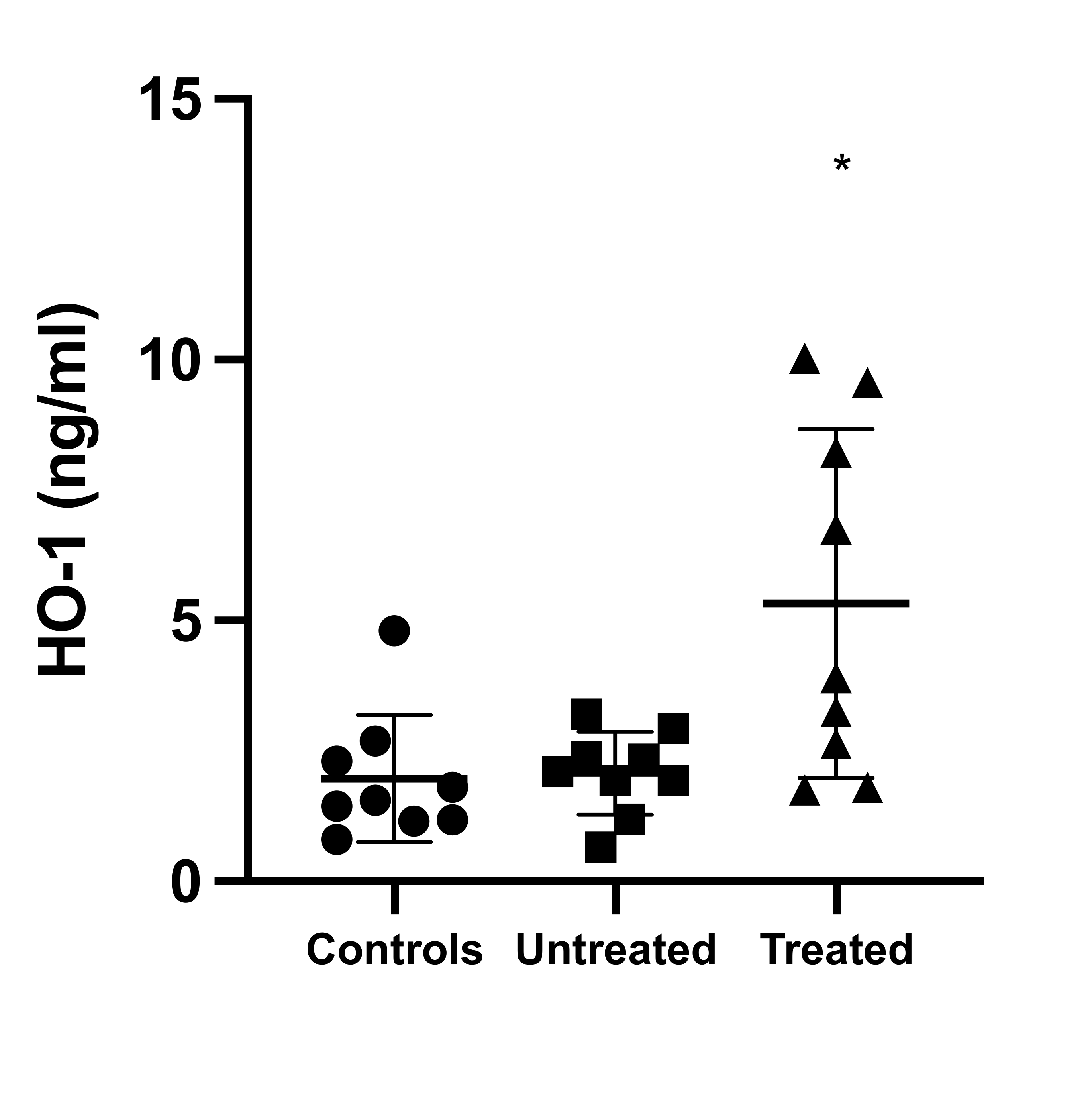
While the impact of OxSt and the effects of tolvaptan on OxSt in ADPKD have been evaluated in vitro and in animal models via the determination of biochemical markers related to OxSt and inflammation [13][14], no human studies are available. To this end, using a molecular biology approach, we evaluated ex vivo the OxSt status of ADPKD patients in our cohort treated with tolvaptan in terms of (1) mononuclear protein expression of p22phox, a subunit of NADPH oxidase key in the transfer of electrons to molecular oxygen to generate superoxide anions and the induction of OxSt [15]; (2) phosphorylation state of MYPT-1, a marker of RhoA/ROCK pathway activity [10][12]; and (3) HO-1 levels, induced and protective against OxSt [16], and compared these to a group of matched untreated ADPKD patients with normal renal function and to a group of healthy subjects.
2. Current Insights
This study showed at the level of molecular biology that OxSt is activated in ADPKD, and that treatment with tolvaptan is associated not only with the reduction in proteins closely related to OxSt signaling, inflammation, and cardiovascular-renal remodeling, but also with the induction of defense/protection toward OxSt. OxSt, in terms of p22phox and MYPT-1 phosphorylation state, was significantly higher in untreated ADPKD patients, whereas it was significantly reduced in tolvaptan-treated ADPKD patients. In addition, the tolvaptan-treated ADPKD patients’ HO-1 levels were significantly higher compared to those of untreated ADPKD patients.
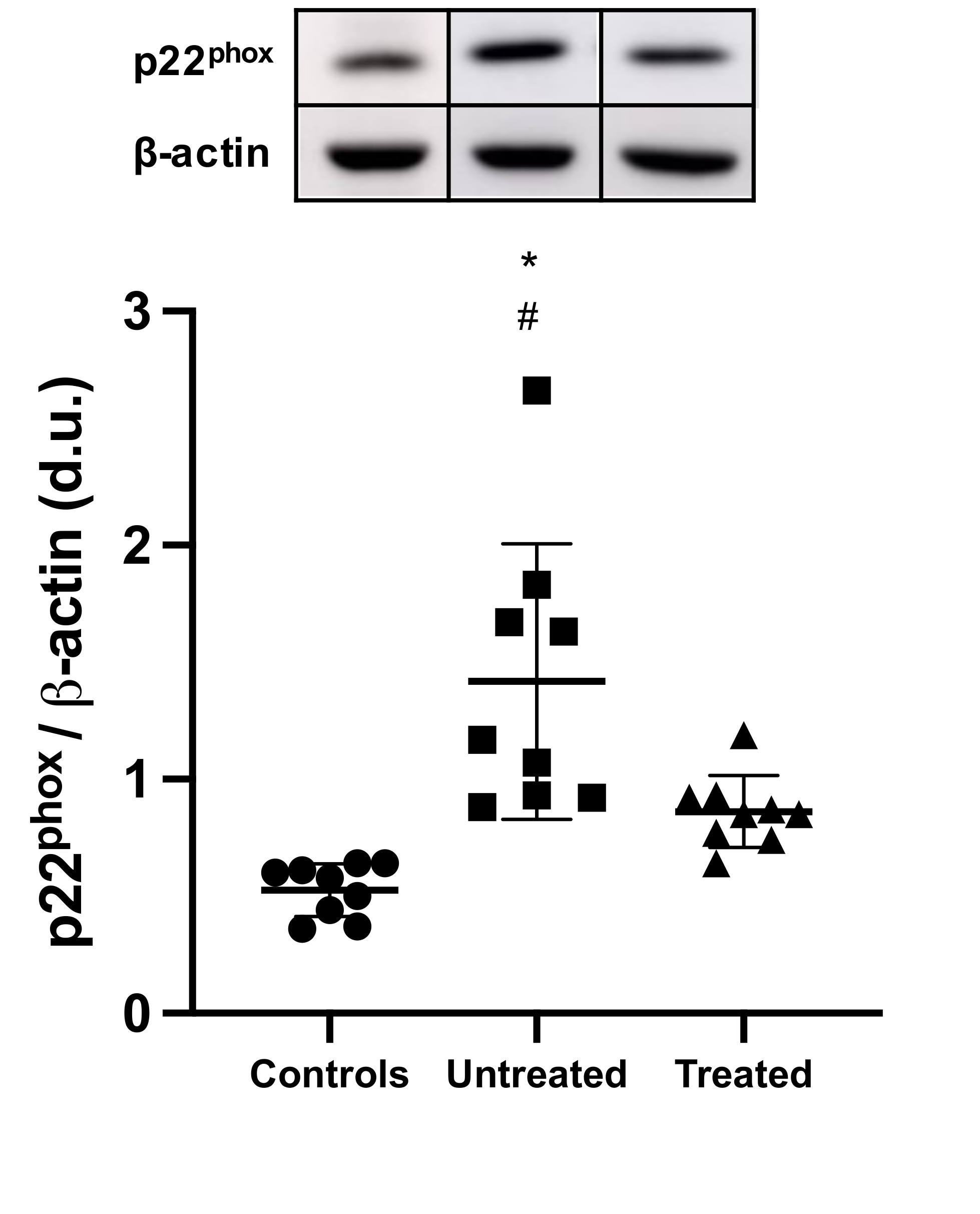
p22phox is a 22 kDa subunit of cytochrome b558 of the NADH/NADPH oxidase present both in leukocytes and in the vascular wall, and is fundamental for the final electron transport from NAD(P)H to heme and molecular oxygen in generating superoxide anions [16]. In tolvaptan-treated ADPKD patients, the reduced p22phox protein levels not only suggests lower OxSt, but also, given its presence in leukocytes, an inhibition of leukocyte activation, which is a well-known cause of OxSt in CKD. As a consequence, this reduction in p22phox protein levels suggests an inhibition of OxSt-mediated signaling mechanisms responsible for vascular remodeling and atherogenesis [5][6].
Another fundamental signaling pathway in OxSt processes is RhoA/ROCK [10][11], a pathway that induces OxSt via upregulation of NADPH oxidase. RhoA/ROCK signal is further involved in the vascular effects of OxSt [10][11]. In addition, it has been recently proven that RhoA/ROCK pathway activation is promoted following the inactivation of PKD-1 via activation of proteins involved in cystogenesis [17][18], whereas ROCK inhibition reduces the activity of proteins involved in cystogenesis, which in turns are upregulated in both PKD1-mutated cystic cells and cystic kidney tissue samples from ADPKD patients [17][18].
We further evaluated heme oxygenase 1 (HO-1), the inducible isoform of HO that protects against oxidative processes. HO-1 acts on heme, producing CO and biliverdin, which is further metabolized to bilirubin, a powerful antioxidant. There are three different isoforms of HO: HO-1, HO-2, and HO-3. HO-1 has a low baseline expression that increases rapidly in the presence of oxidative stress, whereas HO-2 and HO-3 are constitutively expressed [19]. HO-1 mediates production of the vasodilator CO and contributes to the regulation of vascular tone and therefore of blood pressure and endothelial function. Finally, HO-1 has been shown to have a long-term anti-inflammatory and antiproliferative effect [16][20][21].
Tolvaptan is an arginine-vasopressin V2 receptor antagonist and is currently indicated to slow the progression of cyst development and renal failure associated with ADPKD [4]. Ishikawa et al. showed in an animal model that chronic tolvaptan treatment significantly suppressed the upregulation of NADPH oxidase subunits including p22phox, and inhibited RhoA expression, phosphorylation, and MYPT-1 phosphorylation [22]; Novak et al. [23] reported that vascular endothelial cell protein expression of NF-κB was increased in ADPKD, consistent with the presence of local and systemic inflammation. Furthermore, the increased OxSt and inflammation likely contribute to reduce NO bioavailability, as shown in subcutaneous resistance vessels from ADPKD patients where endothelium (NO)-dependent dilatation is impaired. Finally, Fujiki et al.[24] showed that in mpkCCD cells and in the external medulla of mouse kidneys, tolvaptan increased mRNA and protein expressions of HO-1.
The results of our study show for the first time ex vivo in humans that treatment with tolvaptan in ADPKD patients is associated with a reduction of OxSt and an increase of antioxidant defenses such as HO-1. Although our study was performed in a small cohort of patients, in line with the approach from molecular biology, the results support the data obtained by in vitro and animal model studies and provide the mechanistic rationale for the reductions in OxSt and inflammation, as well as protection against the worsening of renal function, endothelial damage, and hypertension in tolvaptan-treated ADPKD patients.
References
- Chapman, A.B.; Devuyst, O.; Eckardt, K.‐U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al.et al. Autosomal‐dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17-27, https://doi.org/10.1038/ki.2015.59.
- Harris, P.C.; Torres, V.E.; Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease.. J. Clin. Investig. 2014, 124, 2315-2324, https://doi.org/10.1172/jci72272..
- Wang, X.; Wu, Y.; Ward, C.J.; Harris, P.C.; Torres, V.E.; Vasopressin Directly Regulates Cyst Growth in Polycystic Kidney Disease.. J. Am. Soc. Nephrol. 2007, 19, 102-108, https://doi.org/10.1681/asn.2007060688.
- Gansevoort, R.T.; Arici, M.; Benzing, T.; Birn, H.; Capasso, G.; Covic, A.; Devuyst, O.; Drechsler, C.; Eckardt, K.‐U.; Emma, F.; et al.et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: A position statement on behalf of the ERA‐EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice.. Nephrol. Dial. Transpl. 2016, 31, 337-348, https://doi.org/10.1093/ndt/gfv456..
- Ravarotto, V.; Simioni, F.; Pagnin, E.; Davis, P.A.; Calò, L.A.; Oxidative stress—Chronic kidney disease—Cardiovascular disease: A vicious circle.. Life Sci. 2018, 210, 125-131, https://doi.org/10.1016/j.lfs.2018.08.067..
- Ravarotto, V.; Bertoldi, G.; Innico, G.; Gobbi, L.; Calò, L.; The Pivotal Role of Oxidative Stress in the Pathophysiology of Cardiovascular‐ Renal Remodeling in Kidney Disease.. Antioxidants 2021, 10, 1041, https://doi.org/10.3390/antiox10071041..
- Calò, L.; Naso, A.; Carraro, G.; Wratten, M.L.; Pagnin, E.; Bertipaglia, L.; Rebeschini, M.; Davis, P.A.; Piccoli, A.; Cascone, C.; et al. Effect of haemodiafiltration with online regeneration of ultrafiltrate on oxidative stress in dialysis patients. Nephrol. Dial. Transpl 2007, 22, 1413–1419, https://doi.org/10.1093/ndt/gfl783.
- Calò, L.A.; Naso, A.; D’Angelo, A.; Pagnin, E.; Zanardo, M.; Puato, M.; Rebeschini, M.; Landini, S.; Feriani, M.; Perego, A.; et al.et al. Molecular Biology‐Based Assessment of Vitamin E‐Coated Dialyzer Effects on Oxidative Stress, Inflammation, and Vascular Remodeling.. Artif. Organs 2011, 35, E33–E39, https://doi.org/10.1111/j.1525‐1594.2010.01125.x..
- Calò, L.A.; Vertolli, U.; Pagnin, E.; Ravarotto, V.; Davis, P.A.; Lupia, M.; Naso, E.; Maiolino, G.; Naso, A.; Increased rho kinase activity in mononuclear cells of dialysis and stage 3–4 chronic kidney disease patients with left ventricular hypertrophy: Cardiovascular risk implications.. Life Sci 2016, 148, 80-85, https://doi.org/10.1016/j.lfs.2016.02.019..
- Calò, L.; Pessina, A.C.; RhoA/Rho‐kinase pathway: Much more than just a modulation of vascular tone. Evidence from studies in humans.. J. Hypertens 2007, 25, 259-264, https://doi.org/10.1097/hjh.0b013e328010d4d2..
- Calò, L.A.; Davis, P.A.; Pagnin, E.; Dal Maso, L.; Maiolino, G.; Seccia, T.M.; Pessina, A.C.; Rossi, G.P.; Increased level of p63RhoGEF and RhoA/Rho kinase activity in hypertensive patients.. J. Hypertens 2014, 32, 331-338, https://doi.org/10.1097/hjh.0000000000000075..
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calò, L.A; ROCK (RhoA/Rho Kinase) in Cardiovascular–Renal Pathophysiology: A Review of New Advancements. J. Clin. Med 2020, 9, 1328, https://doi.org/10.3390/jcm9051328..
- Menon, V.; Rudym, D.; Chandra, P.; Miskulin, D.; Perrone, R.; Sarnak, M.; Inflammation, Oxidative Stress, and Insulin Resistance in Polycystic Kidney Disease.. Clin. J. Am. Soc. Nephrol 2010, 6, 7-13, https://doi.org/10.2215/cjn.04140510..
- Theodorakopoulou, M.; Raptis, V.; Loutradis, C.; Sarafidis, P.; Hypoxia and Endothelial Dysfunction in Autosomal‐Dominant Polycystic Kidney Disease.. Semin. Nephrol 2019, 39, 599-612, https://doi.org/10.1016/j.semnephrol.2019.10.009..
- Griendling, K.K.; Sorescu, D.; Ushio‐Fukai, M.; NAD(P)H oxidase: Role in cardiovascular biology and disease.. Circ. Res 2000, 86, 494-501.
- Immenschuh, S.; Ramadori, G.; Gene regulation of heme oxygenase‐1 as a therapeutic target.. Biochem. Pharmacol 2000, 60, 1121– 1128.
- Cai, J.; Song, X.; Wang, W.; Watnick, T.; Pei, Y.; Qian, F.; Pan, D. A; RhoA–YAP–c‐Myc signaling axis promotes the development of polycystic kidney disease.. Genes Dev 2018, 32, 781-793, https://doi.org/10.1101/gad.315127.118..
- Ma, S.; Guan, K.‐L.; Polycystic kidney disease: A Hippo connection.. Genes Dev. 2018, 32, 737-739, https://doi.org/10.1101/gad.316570.118..
- Nath, K.; Heme oxygenase‐1: A provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 2006, 70, 432-443, https://doi.org/10.1038/sj.ki.5001565..
- Nath, K.A.; Vercellotti, G.M.; Grande, J.P.; Miyoshi, H.; Paya, C.V.; Manivel, J.C.; Haggard, J.J.; Croatt, A.J.; Payne, W.D.; Alam, J.; et al. Heme protein‐induced chronic renal inflammation: Suppressive effect of induced heme oxygenase‐1.. Kidney Int 2001, 59, 106-117.
- Kim, Y.‐M.; Pae, H.‐O.; Park, J.E.; Lee, Y.C.; Woo, J.M.; Kim, N.‐H.; Choi, Y.K.; Lee, B.‐S.; Kim, S.R.; Chung, H.‐T.; et al. Heme Oxygenase in the Regulation of Vascular Biology: From Molecular Mechanisms to Therapeutic Opportunities.. Antioxid. Redox Signal. 2011, 14, 137-167, https://doi.org/10.1089/ars.2010.3153..
- Ishikawa, M.; Kobayashi, N.; Sugiyama, F.; Onoda, S.; Ishimitsu, T.; Renoprotective Effect of Vasopressin V2 Receptor Antagonist Tolvaptan in Dahl Rats with End‐Stage Heart Failure. Int. Hear. J. 2013, 54, 98-106, https://doi.org/10.1536/ihj.54.98..
- Nowak, K.L.; Wang, W.; Farmer‐Bailey, H.; Gitomer, B.; Malaczewski, M.; Klawitter, J.; Jovanovich, A.; Chonchol, M.; Vascular Dysfunction, Oxidative Stress, and Inflammation in Autosomal Dominant Polycystic Kidney Disease.. Clin. J. Am. Soc. Nephrol. 2018, 13, 1493-1501, https://doi.org/10.2215/cjn.05850518..
- Fujiki, T.; Ando, F.; Murakami, K.; Isobe, K.; Mori, T.; Susa, K.; Nomura, N.; Sohara, E.; Rai, T.; Uchida, S.; et al. Tolvaptan activates the Nrf2/HO‐1 antioxidant pathway through PERK phosphorylation.. Sci. Rep 2019, 9, 9245, https://doi.org/10.1038/s41598‐019‐ 45539‐8..
More