The term seizures includes a wide array of different disorders with variable etiology, which currently represent one of the most important classes of neurological illnesses. As a consequence, many different antiepileptic drugs (AEDs) are currently available, exploiting different activity mechanisms and providing different levels of performance in terms of selectivity, safety, and efficacy. AEDs are currently among the psychoactive drugs most frequently involved in therapeutic drug monitoring (TDM) practices. Ezogabine (or retigabine), lacosamide, and zonisamide are the TDM of three AEDs belonging to the class of ion channel agents.
- ezogabine
- lacosamide
- neuroprotection
- retigabine
- zonisamide
- therapeutic drug monitoring
- analytical methods
- sample pretreatment
1. Introduction
The first effective antiepileptic drugs (AEDs) were bromides, introduced in the mid-1850s, and then paraldehyde, in the late 1880s. Since then, many other, vastly more effective and safer drugs have been devised, belonging to many chemical classes, starting from barbiturates in the 1910s, their analogues oxazolidinediones and pyrimidinediones, and hydantoins in the 1930–1960s. Then, benzodiazepines, carboxamides, fatty acids, and their derivatives, fructose derivatives, γ-aminobutyric acid (GABA) derivatives, hydantoins, pyrrolidines, succinimides, sulphonamides, and triazines have been used against seizures [8][2]. Pharmacological activity mechanisms are equally varied, although for some drugs, and entire chemical classes, the precise mechanism is still uncertain [9][3]. The two oldest AEDs, bromide and paraldehyde, are thought to non-selectively depress brain activity; if they have any molecular target, it is still unknown. A large group of AEDs interacts directly with ion channels, acting either as sodium channel blockers (such as carbamazepine, oxcarbazepine, lamotrigine, phenytoin) or as calcium channel blockers (such as ethosuximide); ezogabine seems to act as a potassium channel opener. Many AEDs interact with GABAergic transmission; since GABA is known as the main inhibitory neurotransmitter in the brain, its activation or facilitation is intuitively connected with a possible anticonvulsant effect. Thus, GABA transaminase inhibitors (vigabatrin) and glutamic acid decarboxylase (GAD) inducers (gabapentin) are used as antiepileptic agents. On the other hand, glutamate is generally recognized as the main excitatory brain neurotransmitter, and therefore drugs acting on the glutamatergic system (topiramate) are also known as antiepileptic agents [10][4]. The exact mechanisms of activity of valproic acid, levetiracetam, and stiripentol are still unknown, although of course hypotheses have been proposed [11][5].
2. Ion Channel Agents
2.1. Ezogabine (Retigabine)
3
2.1.1. Therapeutic Drug Monitoring
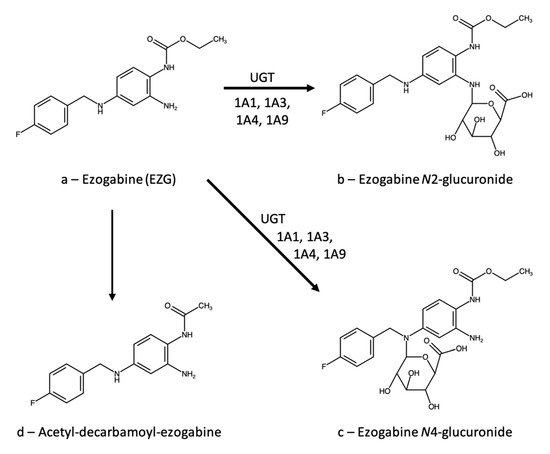
Just a few analytical methods are available for the TDM of EZG; most of them are based on liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) [34,35,36,37][12][13][14][15]. One of these methods [34][12] was applied to evaluate gender differences in EZG pharmacokinetics. Analytical separation was achieved on a C18 column kept at 40 °C with a mobile phase consisting of an aqueous ammonium acetate/methanol/acetonitrile mixture. Sample pretreatment was carried out by liquid–liquid extraction (LLE) of 500 µL of plasma using 5 mL of a diethyl ether/dichloromethane (70/30, v/v) mixture, followed by centrifugation, separation, drying of the organic phase and reconstitution with 100 µL of mobile phase. Detection was performed by MS/MS with triple quadrupole (QqQ) detection. Linearity was achieved in the 10–2000 ng/mL range, with precision RSD values between 1.9% and 9.3%. Extraction yields were in the 88–91% range and accuracy values in the 102–111% range. Stability assays provided recovery values always higher than 80% under all short- and log-term conditions tested. Matrix effect was almost negligible (mean = 3.8%), as was carryover (0.3–0.7% of the limit of quantitation (LOQ) peak area). Instead of a deuterated EZG analogue, oxcarbazepine (Figure 2a) was used as the internal standard (IS), which is obviously a sub-optimal choice. In fact, oxcarbazepine does not bear any close structural resemblance to EZG. Moreover, it is a very popular antiepileptic drug, and this makes its simultaneous administration with EZG quite probable. In these cases, the IS would be totally useless.
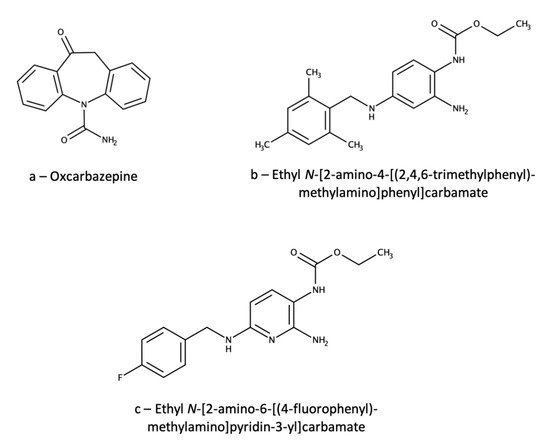
Another method included both EZG and its active N-acetyl metabolite and used automated on-line solid-phase extraction in the form of column-switching [35][13]. In this case, a C8 column specifically suited for basic compounds (LiChrospher RP-Select B) was used, coupled with an acetonitrile/pH 6 ammonium acetate (55/45, v/v) mobile phase. MS/MS detection was carried out by atmospheric pressure chemical ionization (APCI)—QqQ. Sample preparation consisted of a sample dilution step with 800 µL of 13% (v/v) aqueous acetonitrile added to 200 µL of plasma, followed by injection of 500 µL of the mixture into a column-switching system using a preparative C2 column. Interfering matrix components (salts, proteins) were flushed out with 10% (v/v) aqueous acetonitrile, then the analytes were eluted with the mobile phase and switched to the analytical column. In this case, the EZG analogue ethyl N-[2-amino-4-[(2,4,6-trimethylphenyl)methylamino]phenyl]carbamate (Figure 2b) was used as the IS; again, the choice is suboptimal for an LC-MS/MS method, but the structural similarity is quite high.
The third method [36][14] deals with the determination of the N-acetyl metabolite by LC-APCI-MS/MS(QqQ) after pretreatment of 200 µL of plasma by micro-solid phase extraction (µSPE) in 96-well plates with a hydrophilic-lipophilic balance (HLB) sorbent; washing was carried out with two different ammonium acetate/acetonitrile/isopropyl alcohol mixtures, followed by elution with 1.2 mL of acetonitrile/isopropyl alcohol (40/60, v/v). To avoid the possible back-conversion of N-acetyl-EZG glucuronide to the parent N-acetyl-EZG, all materials and reagents for the dilution and µSPE procedures were refrigerated in ice; moreover, the glucuronide was eliminated from the sample by the washing step of the SPE procedure. However, the method only includes N-acetyl-EZG and has only been applied to dog plasma; hence, its usefulness for TDM is almost negligible. The IS was a 13C6-analogue of the analyte, which is a satisfactory choice.
3
2.1.2. Ezogabine Interactions
Although the acetyl EZG metabolite has the capacity of inhibiting the P-glycoprotein (P-gp) transporter, a clinical study has found just a small increase in the digoxin plasma area under the concentration-time curve, which should not have any clinical significance [39][16]. EZG did not have any clinically relevant impact on exposure of hormones in a combined oral contraceptive agent containing norethindrone and ethinyl oestradiol, and the hormones did not alter EZG pharmacokinetic parameters [40][17]. In healthy humans, EZG did not significantly alter the pharmacokinetics of phenobarbital [41][18] and phenobarbitone [42][19].
In humans, ethanol increases exposure to EZG, but this interaction is probably not clinically significant [46][20].
32.1.3. Neuroprotection
3.2. Lacosamide
2.2. Lacosamide
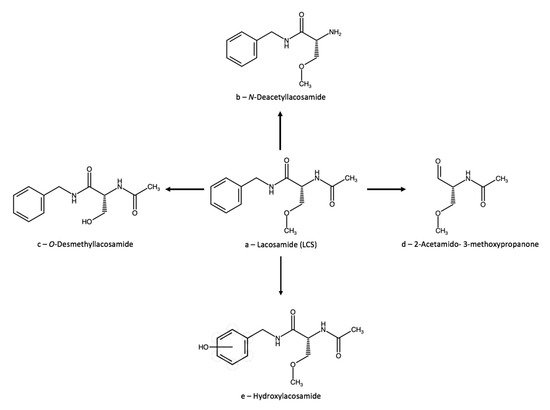
LCS ((2R)-2-acetamido-N-benzyl-3-methoxypropanamide, Figure 3a) is used under the tradename Vimpat for the adjunctive treatment of partial-onset seizures and for neuropathic pain. The therapeutic activity of LCS has been attributed to the inactivation of sodium channels, and in particular to selective, fast binding to the slow inactivated state of Na+ channels [54][23], although slow binding to the fast inactivated state has been alternatively proposed recently [55][24]. It has also been proposed that LCS targets GABAA receptors as well [56][25]. LCS is a chiral compound, but it was traditionally only available as a racemate.3
2.2.1. Therapeutic Drug Monitoring
Single-Drug Methods
3
2.2.2. Lacosamide Interactions
The potential of LCS for interactions, both with other AEDs and with different classes of drugs, seems to be relatively low [78][32].In a retrospective study on 157 drug-resistant patients treated with LCS, levetiracetam resulted to be the compound most frequently associated with LCS in the responder subgroup. This allows us to hypothesize that LCS and levetiracetam can interact synergistically at the pharmacodynamic level [56][25]. Phase I clinical studies have found no significant interactions of LCS with omeprazole [79][33] or digoxin [80][34].
32.2.3. Neuroprotection
3.3. Zonisamide
2.3. Zonisamide
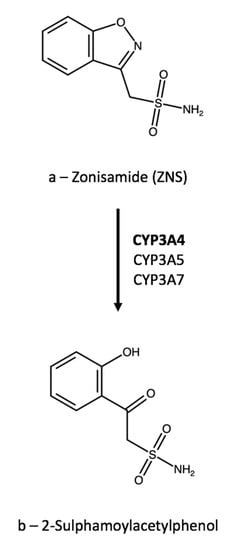
Zonisamide (1,2-benzoxazol-3-ylmethanesulfonamide, ZNS, Figure 65a) is an anticonvulsant agent used primarily for adjunctive treatment of partial seizures. However, it can also be used for a variety of other indications either on- or off-label, including partial and generalized seizures and absence [96][37], bipolar depression as a mood stabilizer [97][38], Parkinson’s disease [98][39], tardive dyskinesia during antipsychotic therapy [99][40], and migraine [100][41]. The mechanism of ZNS antiepileptic activity is still debated, but it is currently reputed to be the blockade of sodium and T-type calcium channels [101][42]. Moreover, the modulation of GABAergic and glutamatergic neurotransmission could have a role in seizure inhibition [102][43]. ZNS is mainly metabolized to 2-sulphamoylacetylphenol (cleavage of the benzisoxazole ring, Figure 65b) by CYP3A4, with minor contributions from CYP3A7 and CYP3A5 [103][44].3
2.3.1. Therapeutic Drug Monitoring
Single-Drug Methods
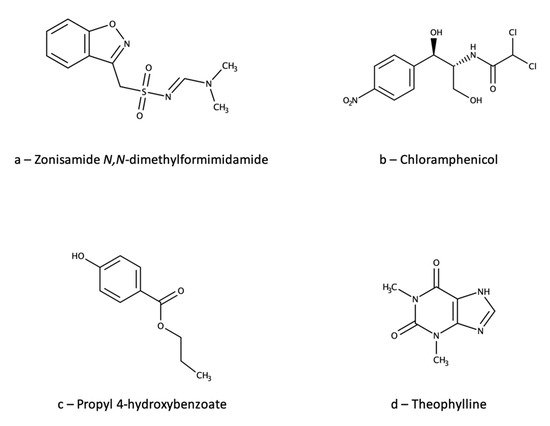
Recently, an HPLC-UV method has been used to verify the reliability of a routine TDM kit based on latex particle-enhanced turbidimetric immunoassay (LTIA) for ZNS [105][45]. The HPLC method used a C18 column and a mobile phase consisting of water, isopropyl alcohol, acetonitrile, and acetic acid. Column temperature was set at 40 °C and detection wavelength at 238 nm. Plasma sample (100 µL) pretreatment was based on a simple PPP with 200 µL of methanol, followed by centrifugation and supernatant injection. The IS was ZNS N,N-dimethylformimidamide (Figure 76a), which is reasonably similar to the analyte; however, the introduction of a relatively big functional group could impair the IS performance and make the method less than ideally reliable. The method was validated for ZNS quantification in the 1–75 µg/mL range, with accuracy in the 93.7–103.7% range and precision RSD values up to 10.3%. The corresponding validation parameters for the LTIA method were: linearity 5–50 µg/mL; accuracy 84.2–106.7; precision RSD ≤ 5.2%. The paper reports strong correlation between the results of the two methods, thus ruling out possible interferences from ZNS metabolites with the LTIA method. However, since the HPLC-UV method did not separate nor analyse any metabolites, and was not validated for selectivity toward them, similar interferences from metabolites in both methods could theoretically not be ruled out.
Multi-Drug Methods
32.3.2. Zonisamide Interactions
Carbamazepine, phenytoin, and phenobarbital all increase ZNS clearance by 24–29%, an interaction that may necessitate a dosage increase [129][60]. Otherwise, ZNS is essentially devoid of clinically significant interactions with other AEDs (such as valproate [130][61] and lamotrigine [131][62]), oral contraceptives, and most other classes of therapeutic agents [132][63]. Conversely, ZNS does not appear to significantly alter the pharmacokinetics of phenytoin [133][64] and carbamazepine [134][65], nor those of oral contraceptives [135][66].
32.3.3. Neuroprotection
References
- World Health Organization. Epilepsy. Available online: http://www.who.int/mediacentre/factsheets/fs999/en/ (accessed on 2 August 2021).
- Aboul–Enein, M.N.; El–Azzouny, A.A.; Saleh, O.A.; Maklad, Y.A. On chemical structures with potent antiepileptic/anticonvulsant profile. Mini Rev. Med. Chem. 2012, 12, 671–700.
- Goldenberg, M.M. Overview of Drugs Used For Epilepsy and Seizures: Etiology; Diagnosis; and Treatment. Pharm. Ther. 2010, 35, 392–415.
- Łukawski, K.; Gryta, P.; Łuszczki, J.; Czuczwar, S.J. Exploring the latest avenues for antiepileptic drug discovery and development. Expert Opin. Drug Dis. 2016, 11, 369–382.
- Lasoñ, W.; Chlebicka, M.; Rejdak, K. Research advances in basic mechanisms of seizures and antiepileptic drug action. Pharmacol. Rep. 2013, 65, 787–801.
- Baftiu, A.; Johannessen Landmark, C.; Nikaj, V.; Neslein, I.-L.; Johannessen, S.I.; Perucca, E. Availability of antiepileptic drugs across Europe. Epilepsia 2015, 56, e191–e197.
- Clark, S.; Antell, A.; Kaufman, K. New antiepileptic medication linked to blue discoloration of the skin and eyes. Ther. Adv. Drug Saf. 2015, 6, 15–19.
- Advance notification of Trobalt® discontinuation. Available online: https://assets.publishing.service.gov.uk/media/57fe4b6640f0b6713800000c/Trobalt_letter.pdf (accessed on 2 August 2021).
- Orhan, G.; Wuttke, T.V.; Nies, A.T.; Schwab, M.; Lerche, H. Retigabine/Ezogabine, a KCNQ/KV7 channel opener: Pharmacological and clinical data. Exp. Opin. Pharmacother. 2012, 13, 1807–1816.
- Hempel, R.; Schupke, H.; McNeilly, P.J.; Heinecke, K.; Kronbach, C.; Grunwald, C.; Zimmermann, G.; Griesinger, C.; Engel, J.; Kronbach, T. Metabolism of retigabine (D–23129), a novel anticonvulsant. Drug Metab. Dispos. 1999, 27, 613–622.
- McNeilly, P.J.; Torchin, C.D.; Anderson, L.W.; Kapetanovic, I.M.; Kupferberg, H.J.; Strong, J.M. In vitro glucuronidation of D–23129, a new anticonvulsant, by human liver microsomes and liver slices. Xenobiotica 1997, 27, 431–441.
- Elkady, E.F.; Aboelwafa, A.A.; Fouad, M.A. Study of gender–related pharmacokinetics of ezogabine in Egyptian volunteers by a validated LC–MS/MS bioanalytical method. J. Adv. Res. 2020, 22, 99–104.
- Knebel, N.G.; Grieb, S.; Leisenheimer, S.; Locher, M. Determination of retigabine and its acetyl metabolite in biological matrices by on–line solid–phase extraction (column switching) liquid chromatography with tandem mass spectrometry. J. Chromatogr. B 2000, 748, 97–111.
- Bu, W.; Nguyen, M.; Xu, C.; Lin, C.-C.; Yeh, L.-T.; Borges, V. Determination of N-acetyl retigabine in dog plasma by LC/MS/MS following off-line μElution 96-well solid phase extraction. J. Chromatogr. B 2007, 852, 465–472.
- Licea Perez, H.; Boram, S.L.; Evans, C.A. Development and validation of a quantitative method for determination of retigabine and its N-acetyl metabolite; overcoming challenges associated with circulating labile N-glucuronide metabolites. Anal. Meth. 2015, 7, 723–735.
- Tompson, D.J.; Crean, C.S.; Buraglio, M.; Arumugham, T. Lack of effect of ezogabine/retigabine on the pharmacokinetics of digoxin in healthy individuals: Results from a drug–drug interaction study. Clin. Pharmacol. 2014, 6, 149–159.
- Crean, C.S.; Tompson, D.J.; Buraglio, M. The effect of ezogabine on the pharmacokinetics of an oral contraceptive agent. Int. J. Clin. Pharmacol. Ther. 2013, 51, 847–853.
- Ferron, G.M.; Patat, A.; Parks, V.; Rolan, P.; Troy, S. Lack of pharmacokinetic interaction between retigabine and phenobarbital at steady–state in healthy subjects. Clin. Pharmacol. Ther. 2001, 69, P48.
- Ferron, G.M.; Patat, A.; Parks, V.; Rolan, P.; Troy, S.M. Lack of pharmacokinetic interaction between retigabine and phenobarbitone at steady–state in healthy subjects. Br. J. Clin. Pharmacol. 2003, 56, 39–45.
- Crean, C.S.; Tompson, D.J. The Effects of Ethanol on the Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Ezogabine (Retigabine). Clin. Ther. 2013, 35, 87–93.
- Nodera, H.; Spieker, A.; Sung, M.; Rutkove, S. Neuroprotective effects of Kv7 channel agonist, retigabine, for cisplatin–induced peripheral neuropathy. Neurosci. Lett. 2011, 505, 223–227.
- Mora, G.; Tapia, R. Effects of retigabine on the neurodegeneration and extracellular glutamate changes induced by 4–aminopyridine in rat hippocampus in vivo. Neurochem. Res. 2005, 30, 1557–1565.
- Peng, Y.-S.; Wu, H.-T.; Lai, Y.-C.; Chen, J.-L.; Yang, Y.-C.; Kuo, C.-C. Inhibition of neuronal Na+ currents by lacosamide: Differential binding affinity and kinetics to different inactivated states. Neuropharmacology 2020, 179, 108266.
- Jo, S.; Bean, B.P. Lacosamide inhibition of Nav1.7 voltage-gated sodium channels: Slow binding to fast-inactivated states. Mol. Pharmacol. 2017, 91, 277–286.
- Limatola, C.; Aronica, E.; Palma, E.; Giallonardo, A.T. A novel action of lacosamide on GABAA currents sets the ground for a synergic interaction with levetiracetam in treatment of epilepsy. Neurobiol. Dis. 2018, 115, 59–68.
- Bentué-Ferrer, D.; Tribut, O.; Verdier, M.-C. Therapeutic drug monitoring of lacosamide Suivi thérapeutique pharmacologique du lacosamide. Therapie 2012, 67, 151–155.
- Schultz, L.; Mahmoud, S.H. Is Therapeutic Drug Monitoring of Lacosamide Needed in Patients with Seizures and Epilepsy? Eur. J. Drug Metab. Pharmacokin. 2020, 45, 315–349.
- May, T.W.; Brandt, C.; Helmer, R.; Bien, C.G.; Cawello, W. Comparison of lacosamide concentrations in cerebrospinal fluid and serum in patients with epilepsy. Epilepsia 2015, 56, 1134–1140.
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, 9–62.
- Sindhu Devi, M.; Peda Varma, D.; Tulja Rani, G.; Srinivas, N. Liquid chromatographic assay of lacosamide in human plasma using liquidliquid extraction. Int. J. Phar. Pharm. Sci. 2014, 6, 530–533.
- Kestelyn, C.; Lastelle, M.; Higuet, N.; Dell’Aiera, S.; Staelens, L.; Boulanger, P.; Boekens, H.; Smith, S. A simple HPLC–UV method for the determination of lacosamide in human plasma. Bioanalysis 2011, 3, 2515–2522.
- Cawello, W.; Stockis, A.; Andreas, J.-O.; Dimova, S. Advances in epilepsy treatment: Lacosamide pharmacokinetic profile. Ann. N.Y. Acad. Sci. 2014, 1329, 18–32.
- Cawello, W.; Mueller-Voessing, C.; Fichtner, A. Pharmacokinetics of lacosamide and omeprazole coadministration in healthy volunteers: Results from a phase I, randomized, crossover trial. Clin. Drug Investig. 2014, 34, 317–325.
- Cawello, W.; Mueller-Voessing, C.; Andreas, J.-O. Effect of lacosamide on the steady-state pharmacokinetics of digoxin: Results from a phase I, multiple-dose, double-blind, randomised, placebo-controlled, crossover trial. Clin. Drug Investig. 2014, 34, 327–334.
- Savran, M.; Ozmen, O.; Erzurumlu, Y.; Savas, H.B.; Asci, S.; Kaynak, M. The Impact of Prophylactic Lacosamide on LPS–Induced Neuroinflammation in Aged Rats. Inflammation 2019, 42, 1913–1924.
- Demiroz, S.; Ur, K.; Ulucan, A.; Bengu, A.S.; Demiralin Ur, F.; Gergin, O.O.; Erdem, S. Neuroprotective effects of lacosamide in experimental traumatic spinal cord injury in rats. Turk. Neurosurg. 2019, 29, 718–723.
- Excegran Tablets 100 mg & Excegran Powder 20%. Available online: https://web.archive.org/web/20070928063802/http://www.e-search.ne.jp/~jpr/PDF/DAINIP03.PDF (accessed on 2 August 2021).
- Zonegran. Available online: http://www.bellaireneurology.com/seizure/epil_trt_zonegran.html (accessed on 2 August 2021).
- Grover, N.D.; Limaye, R.P.; Gokhale, D.V.; Patil, T.R. Zonisamide: A review of the clinical and experimental evidence for its use in Parkinson’s disease. Indian J. Pharmacol. 2013, 45, 547–555.
- Iwata, Y.; Irie, S.; Uchida, H.; Suzuki, T.; Watanabe, K.; Iwashita, S.; Mimura, M. Effects of zonisamide on tardive dyskinesia: A preliminary open–label trial. J. Neurol. Sci. 2012, 315, 137–140.
- Zonisamide. Martindale: The Complete Drug Reference; Brayfield, A., Ed.; MedicinesComplete: London, UK, 2016.
- Leppik, I.E. Zonisamide: Chemistry, mechanism of action, and pharmacokinetics. Seizure 2004, 13 (Suppl. 1), S5–S10.
- Ueda, Y.; Doi, T.; Tokumaru, J.; Willmore, L.J. Effect of zonisamide on molecular regulation of glutamate and GABA transporter proteins during epileptogenesis in rats with hippocampal seizures. Brain Res. Mol. 2003, 116, 1–6.
- Stiff, D.D.; Robicheau, J.T.; Zemaitis, M.A. Reductive metabolism of the anticonvulsant agent zonisamide, a 1,2–benzisoxazole derivative. Xenobiotica 1992, 22, 1–11.
- Eto, D.; Tanaka, R.; Suzuki, Y.; Sato, Y.; Itoh, H. Comparison of performance characteristics between high–performance liquid chromatography and latex agglutination turbidimetric immunoassay for therapeutic drug monitoring of zonisamide. J. Clin. Lab. Anal. 2019, 33, e22940.
- Eto, D.; Tanaka, R.; Suzuki, Y.; Sato, Y.; Itoh, H. Comparison of performance characteristics between high–performance liquid chromatography and latex agglutination turbidimetric immunoassay for therapeutic drug monitoring of zonisamide. J. Clin. Lab. Anal. 2019, 33, e22940.
- Lourenço, D.; Sarraguça, M.; Alves, G.; Coutinho, P.; Araujo, A.R.T.S.; Rodrigues, M. A novel HPLC method for the determination of zonisamide in human plasma using microextraction by packed sorbent optimised by experimental design. Anal. Meth. 2017, 9, 5910–5919.
- Makino, K.; Goto, Y.; Sueyasu, M.; Futagami, K.; Kataoka, Y.; Oishi, R. Micellar electrokinetic capillary chromatography for therapeutic drug monitoring of zonisamide. J. Chromatogr. B 1997, 695, 417–425.
- Munshi, R.P.; Gawde, N. Development and Validation of a HPTLC Method to Determine Serum Zonisamide levels for Therapeutic Drug Monitoring in Clinical Settings. Indian J. Pharm. Sci.
- Protti, M.; Sberna, P.M.; Sberna, A.E.; Ferrante, R.; Mandrioli, R.; Mercolini, L. Enhanced urinary stability of peptide hormones and growth factors by dried urine microsampling. J. Pharm. Biomed. Anal. 2021, 204, 114234.
- Mercolini, L.; Mandrioli, R.; Protti, M. Quantitative microsampling for bioanalytical applications related to the SARS–CoV–2 pandemic: Usefulness, benefits and pitfalls. J. Pharm. Biomed. Anal. 2020, 191, 113597.
- Protti, M.; Marasca, C.; Cirrincione, M.; Mandrioli, R.; Cavalli, A.; Mercolini, L. Assessment of capillary volumetric blood microsampling for the analysis of central nervous system drugs and metabolites. Analyst 2020, 145, 5744–5753.
- Marasca, C.; Protti, M.; Mandrioli, R.; Atti, A.R.; Armirotti, A.; Cavalli, A.; De Ronchi, D.; Mercolini, L. Whole blood and oral fluid microsampling for the monitoring of patients under treatment with antidepressant drugs. J. Pharm. Biomed. Anal. 2020, 118, 113384.
- Vincze, I.; Rudge, J.; Vásárhelyi, B.; Karvaly, G.B. Analysis of 14 drugs in dried blood microsamples in a single workflow using whole blood and serum calibrators. Bioanalysis 2020, 12, 1243–1261.
- Mandrioli, R.; Mercolini, L.; Protti, M. Blood and plasma volumetric absorptive microsampling (VAMS) coupled to LC–MS/MS for the forensic assessment of cocaine consumption. Molecules 2020, 25, 1046.
- Mercolini, L.; Mandrioli, R.; Protti, M. Tutorial: Volumetric absorptive microsampling (VAMS). Anal. Chim. Acta 2019, 1046, 32–47.
- Protti, M.; Sberna, P.M.; Sardella, R.; Vovk, T.; Mercolini, L.; Mandrioli, R. VAMS and StAGE as innovative tools for the enantioselective determination of clenbuterol in urine by LC–MS/MS. J. Pharm. Biomed. Anal. 2021, 195, 113873.
- D’Urso, A.; Cangemi, G.; Barco, S.; Striano, P.; D’avolio, A.; De Grazia, U. LC-MS/MS-Based Quantification of 9 Antiepileptic Drugs From a Dried Sample Spot Device. Ther. Drug Monit. 2019, 41, 331–339.
- Baldelli, S.; Cattaneo, D.; Giodini, L.; Baietto, L.; Di Perri, G.; D’Avolio, A.; Clementi, E. Development and validation of a HPLC-UV method for the quantification of antiepileptic drugs in dried plasma spots. Clin. Chem. Lab. Med. 2015, 53, 435–444.
- Okada, Y.; Seo, T.; Ishitsu, T.; Wanibuchi, A.; Hashimoto, N.; Higa, Y.; Nakagawa, K. Population estimation regarding the effects of cytochrome P450 2C19 and 3A5 polymorphisms on zonisamide clearance. Ther. Drug Monit. 2008, 30, 540–543.
- Ragueneau-Majlessi, I.; Levy, R.H.; Brodie, M.; Smith, D.; Shah, J.; Grundy, J.S. Lack of pharmacokinetic interactions between steady-state zonisamide and valproic acid in patients with epilepsy. Clin. Pharmacokin. 2005, 44, 517–523.
- Levy, R.H.; Ragueneau-Majlessi, I.; Brodie, M.J.; Smith, D.F.; Shah, J.; Pan, W.-J. Lack of clinically significant pharmacokinetic interactions between zonisamide and lamotrigine at steady state in patients with epilepsy. Ther. Drug Monit. 2005, 27, 193–198.
- Sills, G.J.; Brodie, M.J. Pharmacokinetics and drug interactions with zonisamide. Epilepsia 2007, 48, 435–441.
- Levy, R.H.; Ragueneau-Majlessi, I.; Garnett, W.R.; Schmerler, M.; Rosenfeld, W.; Shah, J.; Pan, W.-J. Lack of a clinically significant effect of zonisamide on phenytoin steady-state pharmacokinetics in patients with epilepsy. J. Clin. Pharmacol. 2004, 44, 1230–1234.
- Ragueneau-Majlessi, I.; Levy, R.H.; Bergen, D.; Garnett, W.; Rosenfeld, W.; Mather, G.; Shah, J.; Grundy, J.S. Carbamazepine pharmacokinetics are not affected by zonisamide: In vitro mechanistic study and in vivo clinical study in epileptic patients. Epilepsy Res. 2004, 62, 1–11.
- Griffith, S.G.; Dai, Y. Effect of zonisamide on the pharmacokinetics and pharmacodynamics of a combination ethinyl estradiol–norethindrone oral contraceptive in healthy women. Clin. Ther. 2004, 26, 2056–2065.
- Hossain, M.M.; Weig, B.; Reuhl, K.; Gearing, M.; Wu, L.J.; Richardson, J.R. The anti–parkinsonian drug zonisamide reduces neuroinflammation: Role of microglial Nav 1.6. Exp. Neurol. 2018, 308, 111–119.
- Sano, H.; Murata, M.; Nambu, A. Zonisamide reduces nigrostriatal dopaminergic neurodegeneration in a mouse genetic model of Parkinson’s disease. J. Neurochem. 2015, 134, 371–381.
- Asanuma, M.; Miyazaki, I.; Diaz-Corrales, F.J.; Kimoto, N.; Kikkawa, Y.; Takeshima, M.; Miyoshi, K.; Murata, M. Neuroprotective effects of zonisamide target astrocyte. Ann. Neurol. 2010, 67, 239–249.