The nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that acts through antioxidant-responsive elements (ARE) in the promoter region of deoxyribonucleic acid (DNA) and it transcriptionally promotes the expression of Phase 2 metabolism/antioxidant enzymes, such as glutathione S-transferase (GST), nicotinamide adenine dinucleotide (phosphate) (NAD(P)H) quinone oxidoreductase-1 (NQO-1), and heme oxygenase-1 (HO-1). The Nrf2-mediated pathway is now recognized to occupy a central position in cell defense mechanisms. Here, we discuss the historical discovery of Nrf2 and the regulatory mechanism of the Nrf2-mediated pathway, focusing on the interacting proteins and post-translational modifications.
- nuclear factor erythroid 2-related factor 2 (Nrf2)
- antioxidant responsive elements (ARE)
- kelch-like ECH-associated protein 1 (Keap1)
- b-transducin repeat-containing protein (b-TrCP)
- phosphorylation
- acetylation
1. Discovery the Nrf2/ARE-Mediated Cell Defense Mechanism
In 1988–1989, Telakowski-Hopkins et al. and Daniel et al. reported that the 5’-flanking region of a rat or mouse GST Ya subunit gene contains two cis-acting regulatory elements, one of which is required for constitutive expression, and the other is required for inducible expression in response to planar aromatic compounds, such as β-naphthoflavone [1][2]. The cis-acting element required for inducible expression was responsive to planar aromatic compounds only in cells with a functional aryl hydrocarbon receptors (AhR) and cytochrome P1-450 activity, whereas it directly responded to electrophilic inducers such as trans-4-phenyl-3-buten-2-one, dimethyl fumarate, and t-butylhydroquinone, thus the element was designated as electrophile-responsive elements (EpRE) by Friling et al. in 1990 [3].
In 1990, Rushmore et al. identified a xenobiotic-responsive element in the 5’ flanking sequence of a GST Ya subunit gene partly responsible for the basal level as well as inducible expression of the Ya subunit gene by planar aromatic compounds, such as β-naphthoflavone [4]. The element was directly responsive to phenolic antioxidants, such as t-butylhydroquinone, and it was named antioxidant-responsive elements (ARE) [5]. Mutational and deletion analyses of the GST Ya subunit gene and the NQO-1 gene identified the core sequence of the ARE; 5′-puGTGACNNNGC-3′, 3′-pyCACTGNNNCG-5′, where N is any nucleotide [6]. ARE and EpRE appeared to be the identical deoxyribonucleic acid (DNA) sequences similar to activator protein-1 (AP-1) binding sites [7].
A tandem-repeated consensus sequence is present in the β-globin locus control region and binds to the AP-1 family and/or to the nuclear factor erythroid 2 (NF-E2) [8]. In 1993, Dr. Kan’s research team screened a human erythroleukemia cell line (K562) complementary DNA (cDNA) library using the tandem repeat as a recognition site probe, and isolated several cDNAs, two of which had remarkable similarities with the genes encoding NF-E2, thus the novel gene products were named Nrf1 and Nrf2 [9][10]. Nrf1 and Nrf2 genes encode members of the basic leucine zipper (b-zip) protein family that activate transcription via the DNA binding domain highly homologous to that of NF-E2. A high degree of homology is found in the b-zip and neighboring regions among Nrf2, Nrf1, NF-E2, and the Drosophila segmentation protein Cap ‘N’ Collar (CNC) [9][10]. Nrf1 and Nrf2 are different in molecular size and overall structure but share several homology domains [11].
Chan et al. reported that Nrf2 was not essential for growth, development, or erythropoiesis in mammalian cells based on the observation in Nrf2 knockout mice [12], but a role of Nrf2 in the regulation of hematopoietic stem cell function was substantiated later by Tsai et al. [13].
2. Keap1-Dependent Regulation of Nrf2
After AREs had been known to be involved in the transcriptional control of a group of phase II detoxifying enzymes, the identity of Nrf2 as a transcription factor that acts through AREs was revealed by Itoh et al. using homozygous Nrf2-mutant mice [14]. Unlike Nrf2 knockout mice, Nrf1 knockout mice displayed anemia due to impaired fetal liver erythropoiesis and embryonic lethality [15]. Nrf1 upregulated a unique battery of ARE-dependent genes, such as metallothionein (MT)-1 and -2 in addition to typical Nrf2 target genes, such as NQO-1 [16]. Nrf3 was also recognized in 1999 [17]. Nrf3 knockout mice displayed no overt phenotype [18]. All Nrf members, such as Nrf1, -2, and -3, regulate the expression of ARE-dependent genes, with differential and overlapping DNA-binding and transcriptional activities, and they show differed tissue distribution [19].
In 1999, Itoh et al. observed that the deletion of Nrf2-ECH (erythroid cell-derived protein with CNC homology) homology (Neh) 2 domain of Nrf2 led to more potent transactivation in erythroblasts, indicating that Nrf2 activity is normally repressed and the Neh2 domain is involved in the negative regulation. Using the yeast two-hybrid system, they discovered a novel cytoplasmic protein that binds to Nrf2 through the Neh2 domain, and negatively regulates the transactivation potential of Nrf2, which was named Kelch-like ECH-associated protein 1 (Keap1) [20] because of its structural similarity to a Drosophila actin binding protein called Kelch [21] and chicken ECH [22]. They further found that Nrf2 bound to Keap1 is rapidly degraded through the proteasome pathway, while electrophiles can liberate Nrf2 from Keap1, and cause Nrf2 nuclear translocation with concomitant stabilization [23].
In 2002, Zipper et al. showed that the two Keap1 molecules form a homodimer via their Broad-Complex, Tramtrack and Bric-a-Brac (BTB) domains, and Keap1 dimerization is required for Nrf2 sequestration and transcriptional repression [24]. Dinkova-Kostova et al. proposed that the modification of sulfhydryl groups of certain cysteine (Cys, C) residues in Keap1, rather than Nrf2, may be important to the regulation of the Nrf2 pathway, as the Neh2 region of Nrf2 contains no Cys residues [25]. A number of Cys residues in the intervening region (IVR) between the BTB domain and Kelch-repeats of the Keap1 protein, such as Cys257, Cys273, Cys288, and Cys297, were found to be particularly reactive [25].
In 2003, Zhang et al. identified redox-sensitive Cys residues in Keap1, including Cys151, that were required for Keap1-dependent ubiquitylation of Nrf2 and degradation by proteasomes [26]. They proposed that Keap1 is a redox-regulated substrate adaptor protein for a cullin (Cul) 3-dependent E3 ubiquitin ligase complex that is specifically targeted for inhibition by oxidative stress [27].
The Neh2 domain of Nrf2 has both 29DLG31 and 79ETGE82 motifs [28][29], one of which motifs binds to one molecule of Keap1, and the other binds to a second molecule of Keap1 [30], bringing Nrf2 to the proximity of Cul3, a scaffold protein that forms the E3 ligase complex with really interesting new gene (RING)-box 1 (RBX1) [31][32][33]. Under the basal, unstressed condition, the Cul3 complex ubiquitylates Nrf2 at a lysine (Lys, K) residing between the 29DLG31 and 79ETGE82 motifs [30].
In 2005, Velichkova et al. showed that Keap1 sequestered Nrf2 in the cytoplasm through an active chromosome region maintenance 1 (Crm1)/exportin-1-dependent nuclear export mechanism [34]. They identified a nuclear export signal (NES) consensus sequence in the IVR of Keap1, and mutation of hydrophobic amino acids in the NES sequence resulted in nuclear accumulation of Keap1 and Nrf2, as did leptomycin B, which inactivates Crm1. Thus, a modification of Keap1’s NES was proposed to promote the entry of both Keap1 and Nrf2 into the nucleus and transcriptional transactivation of ARE-driven genes.
Figure 1 shows the domain structures of Keap1 and Nrf2.
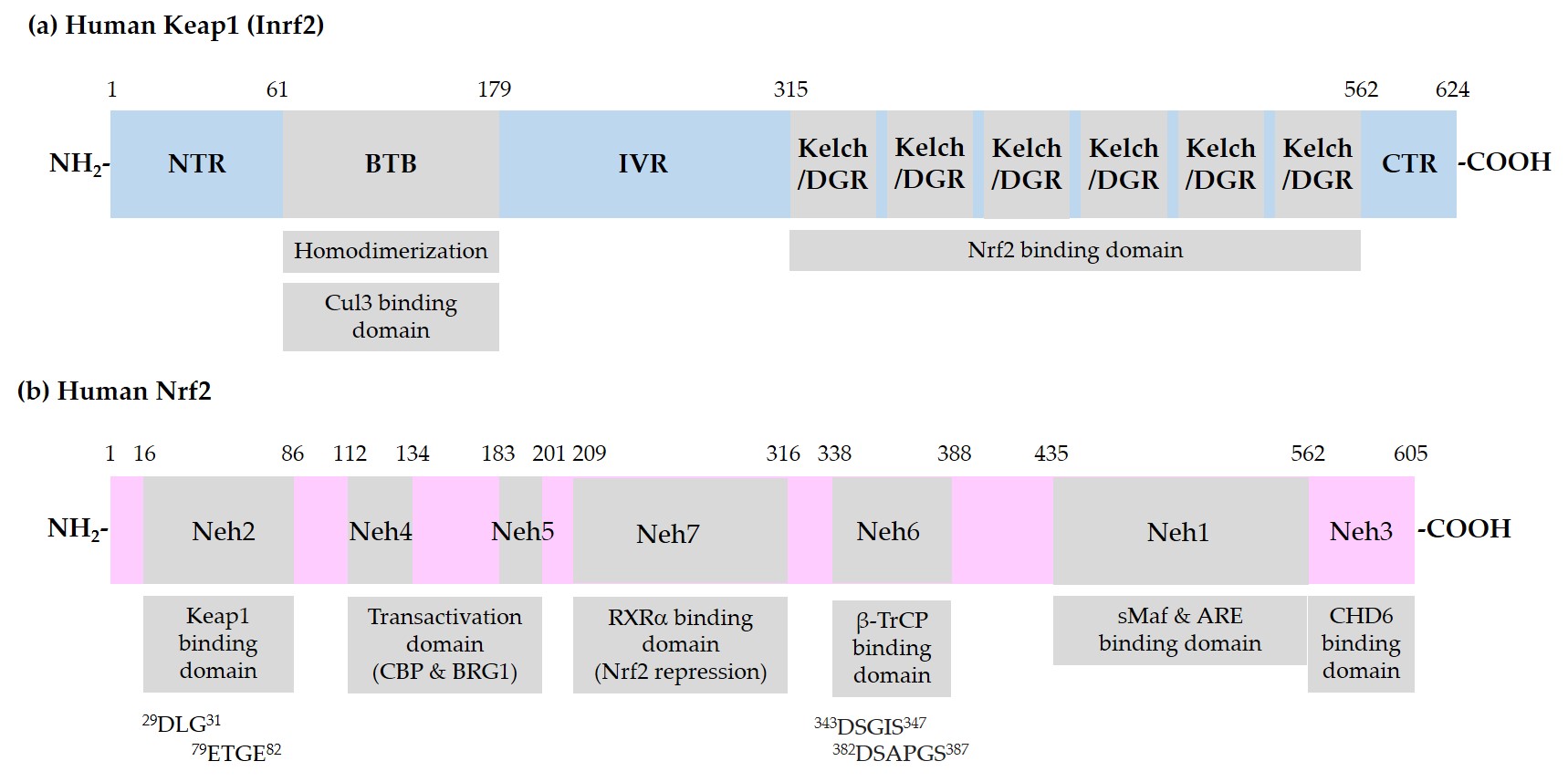
Figure 1. Schematic representation of domain structures of Kelch-like ECH-associated protein 1 (Keap1) and nuclear factor erythroid 2-related factor 2 (Nrf2). (a) Functional domains of Keap1: N-terminal region (NTR), Broad-Complex, Tramtrack, and Bric-a-Brac (BTB) domain, intervening region (IVR), Kelch/Double glycine repeats (DGR) domain, and C-terminal region (CTR). BTB is responsible for homodimerization between two Keap1 molecules and association with a Cullin (Cul) 3/E3 ubiquitin ligase complex. IVR contains reactive cysteine (Cys) residues. Kelch/DGR domain is responsible for binding to 29DLG31 and 79ETGE82 motifs in the Nrf2-ECH homology (Neh) 2 domain of Nrf2; (b) Neh domains of Nrf2: Neh1 domain is the binding site for small musculoaponeurotic fibrosarcoma (sMaf) proteins and antioxidant response element (ARE) in the promoter region of deoxyribonucleic acid (DNA). Neh2 domain contains 29DLG31 and 79ETGE82 motifs, through which Nrf2 interacts with Kelch/DGR domains of two Keap1 molecules of a homodimer. Neh6 domain contains 343DSGIS347 and 382DSAPGS387 motifs, through which Nrf2 interacts with β-transducin repeat-containing protein (β-TrCP). Neh4 and -5 domains are involved in transcriptional activation. Binding domain for cAMP-responsive element binding protein (CREB) binding protein (CBP), Brahma-related gene 1 (BRG1), chromo-ATPase/helicase DNA binding protein (CHD6), and retinoic X receptor α (RXRα) are indicated.
Figure 2 illustrates a schematic model for sequestration of Nrf2 by a Keap1/Cul3/Rbx1/E3 ubiquitin ligase complex and subsequent ubiquitylation for proteasomal degradation of Nrf2.
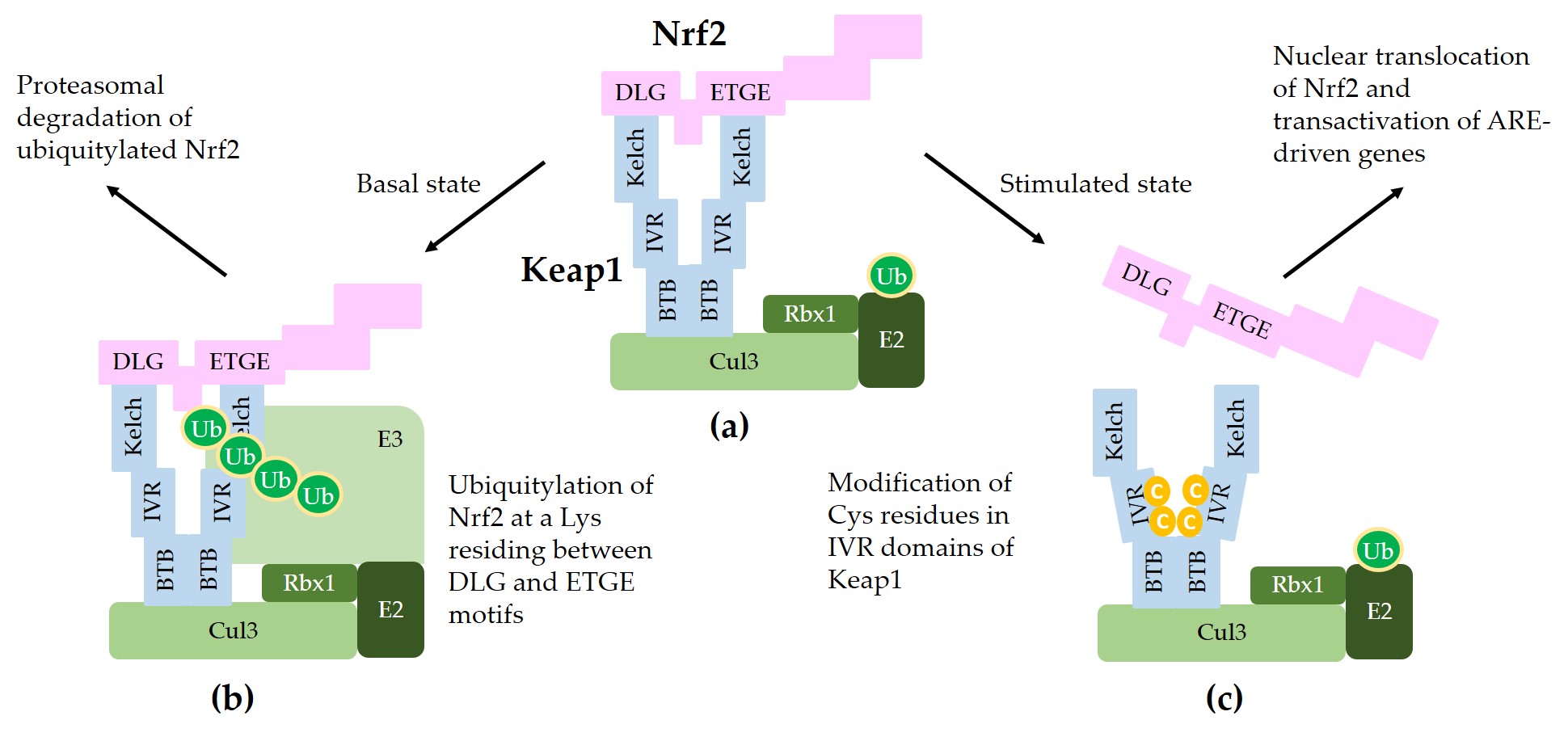
Figure 2. A model for interaction between Nrf2 and Keap1 in the canonical Nrf2 pathway. (a) Two molecules of Keap1 form a homodimer through the BTB domain and bind to Cul3. Dimeric keap1 molecules capture a Nrf2 molecule through the Kelch/DGR domains that interact with the 29DLG31 and 79ETGE82 motifs of Nrf2, resulting in the sequestration of Nrf2 to a Cul3/really interesting new gene (RING)-box 1 (RBX1)/E3 ubiquitin ligase complex. (b) Under basal, unstressed conditions, the Cul3 complex ubiquitylates Nrf2 at a lysine (Lys, K) residue between the 29DLG31 and 79ETGE82 motifs. Ubiquitylated Nrf2 is then transferred to the 26S proteasomes, where it is degraded. (c) When cells are stimulated by oxidants or xenobiotics, certain cysteine (Cys, C) residues of Keap1 can be modified, thereby preventing ubiquitylation and releasing Nrf2 protein. The released Nrf2 can translocate to the nucleus and get involved in the transcriptional activation of ARE resulting in the enhanced gene expression of target genes.
3. Other Proteins that Interact with Nrf2 or ARE
In 1994, Igarashi et al. showed that the small musculoaponeurotic fibrosarcoma (sMaf) proteins, which possess a b-zip DNA binding domain but lack a canonical transactivation domain, directly associate with Nrf2 that also contains a b-zip domain, conferring DNA binding property. Heterodimers of sMaf and Nrf2 induced active transcription whereas homodimers of sMaf molecules act as negative regulators, indicating that positive and negative regulation of transcription can be achieved by controlling the relative concentrations of Nrf2 and sMaf in the nucleus [35].
In 2001, Katoh et al. found that two transcription activation domains of Nrf2, Neh4 and Neh5, cooperatively bind to a transcriptional coactivator, cyclic AMP-responsive element binding protein (CREB) binding protein (CBP) [36]. In 2007, Zhang et al. showed that binding of CBP and Brahma-related gene 1 (BRG1) to Neh5 domain of Nrf2 enhanced HO-1 promoter activity cooperatively [37].
In 2005, Dhakshinamoorthy et al. demonstrated that BTB domain and CNC homolog 1 (Bach1) binds to ARE as a heterodimer with sMaf proteins but not as a homodimer or heterodimer with Nrf2, and thereby competes with Nrf2 leading to the suppression of ARE-mediated NQO-1 gene expression [38]. Nioi et al. found that the Neh3 domain of Nrf2 is needed for its interaction with chromo-ATPase/helicase DNA binding protein 6 (CHD6) to induce transcriptional activity, regulating NQO-1 gene expression [39].
In 2013, Wang et al. reported a function of retinoic X receptor α (RXRα) as a Nrf2 repressor. They observed that RXRα physically interacts with Nrf2 and binds to ARE sequences in the promoters of Nrf2-regulated genes, and suggested a hypothesis that a direct interaction between Nrf2 and RXRα on gene promoters accounts for the antagonism of ARE-driven gene expression. They designated the Neh7 domain, comprising amino acids 209–316 in human Nrf2, for interaction with the DNA binding domain of RXRα.
In 2012, Rojo et al. observed that a cancer-chemopreventive agent, nordihydroguaiaretic acid (NDGA), increased the level of Nrf2 protein and expression of HO-1 in wild-type mouse embryo fibroblasts (MEFs) and in Keap1(-/-) MEFs, but not in Nrf2(-/-) MEFs, implying that Keap1-independent mechanisms regulate Nrf2 stability and HO-1 induction [40]. NDGA caused inhibitory phosphorylation of glycogen synthase kinase 3β (GSK3β), and this was associated with a reduction in Neh6 phosphorylation. Subsequently, two serine (Ser, S) residues, Ser344 and Ser347 (Mouse Ser335 and Ser338) in the Neh6 domain of Nrf2, were identified to be phosphorylated by GSK3 [41].
In 2013, Chowdhry et al. proved that Nrf2 can be ubiquitylated by a β-transducin repeat-containing protein (β-TrCP)/S-Phase kinase-associated protein (Skp) 1/Cul1/Rbx1/E3 ubiquitin ligase complex [42]. In this complex, β-TrCP acts as a substrate receptor and captures Nrf2 through binding to 343DSGIS347 and 382DSAPGS387 motifs that are present in the Neh6 domain of Nrf2. GSK3-mediated phosphorylation of Ser residues in the 343DSGIS347 motif enhanced the interaction between Nrf2 and β-TrCP, which resulted in increased ubiquitylation of Nrf2 and subsequent proteasomal degradation. As the function of β-TrCP in the regulation of Nrf2 stability is distinct from Keap2, Dr. Cuadrado has proposed “Dual nuclear flux control model for regulation of Nrf2 stability by Keap1” by defining the roles of Keap1and β-TrCP as a “limiter valve” and a “regulator valve,” respectively, to control the nuclear flux of Nrf2 [43].
There are many other proteins that are involved in the regulation of the Nrf2 pathway, such as the endoplasmic reticulum (ER)-associated E3 ubiquitin ligase, 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase degradation 1 (HRD1), that interacts with Nrf2 through Neh4/5 domains and promotes the proteasomal degradation of Nrf2 [44], and the scaffold protein Caveolin 1 that was proposed to compete with Keap1 for Nrf2 binding, thereby enhancing the stability of Nrf2 [45], but all cannot be covered here due to my limited capability.
Figure 3 illustrates the interaction of Nrf2 with various proteins that are known to regulate the sequestration of Nrf2 or its transcriptional activity.
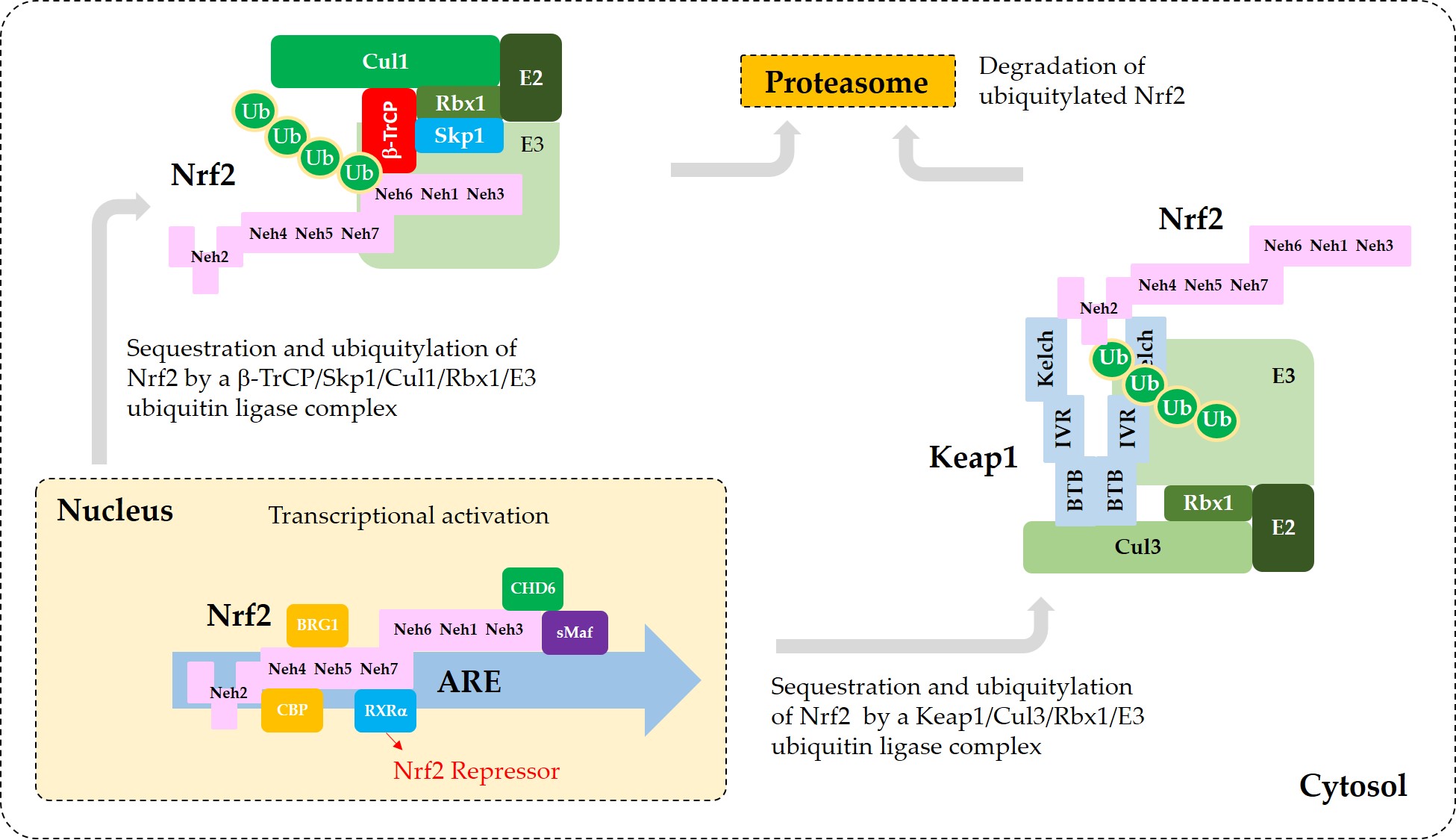
Figure 3. A model for the interaction of Nrf2 with various proteins that promote sequestration of Nrf2 or enhance or suppress its transcriptional activity. In the nucleus, heterodimerization of Nrf2 with sMaf through its Neh1 domain is required for efficient binding to the ARE in the promoter region of DNA. Binding of CBP and BRG1 at Neh4/5 domains and CHD6 at Neh3 domain of Nrf2 are required for full transcriptional activity of Nrf2. RXRα binds to Neh7 domain of Nrf2, acting as a repressor. In the cytosol, Keap1 captures Nrf2 through binding to 29DLG31 and 79ETGE82 motifs in Neh2 domain and sequesters it to a Cul3/Rbx1/E3 ubiquitin ligase complex. Alternatively, β-TrCP captures Nrf2 through binding to 343DSGIS347 and 382DSAPGS387 motifs in Neh6 domain and sequesters it to a Cul1/Skp1/Rbx1/E3 ubiquitin ligase complex. Ubiquitylated Nrf2 is then transferred to the 26S proteasomes, where it is degraded.
4. Phosphorylation of Nrf2
In 2002, Huang et al. found that Nrf2 could be phosphorylated at the Ser40 residue by protein kinase C (PKC), and the phosphorylation enhanced ARE-mediated transcription activity [46]. They further showed that Nrf2 was degraded by the ubiquitin-dependent pathway and that phosphorylation of Nrf2-Ser40 leads to an increase in its stability and subsequent transactivation activity [47]. In 2003, Numazawa et al. proposed that atypical PKC-iota (i) is responsible for phosphorylation of Nrf2-Ser40 [48]. Cullinan et al. demonstrated that Nrf2 is a substrate of protein kinase RNA-activated (PKR)-like ER kinase (PERK). PERK-dependent phosphorylation of Nrf2 at the N-terminal region was shown to trigger dissociation of Nrf2/Keap1 complexes and subsequent Nrf2 nuclear import [49].
In 2006, Jain et al. showed that Fyn kinase can phosphorylate tyrosine (Tyr, Y) 576 (Mouse Tyr568) in the NES of Nrf2, and this phosphorylation is required for Crm1-mediated nuclear export and subsequent degradation of Nrf2 [50]. They further demonstrated that GSK3β acts as an upstream regulator of Fyn kinase in the control of the nuclear export of Nrf2 [51]. In this study, hydrogen peroxide (H2O2) activated GSK3β by phosphorylation of Tyr216 residue and the activated GSK3β phosphorylated Fyn kinase at threonine (Thr, T) residues, causing nuclear accumulation of Fyn. Fyn-dependent phosphorylation of Tyr576 (Mouse Tyr568) of Nrf2 led to nuclear export, ubiquitylation, and degradation of Nrf2.
Salazar et al. showed that GSK3β can directly phosphorylate Nrf2 and induce exclusion of Nrf2 from the nucleus [52]. Phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB, Akt) increased the nuclear translocation of Nrf2 by inhibiting GSK3β kinase activity. In 2012, Rada et al. identified Ser344 and Ser347 (Mouse Ser335 and Ser338) of Nrf2 as the GSK3β-mediated phosphorylation sites [41]. These Ser residues are contained in one of the two binding motifs for β-TrCP.
Of the mitogen-activated protein kinases (MAPKs), p38 was first shown to phosphorylate Nrf2 and promote the association between Nrf2 and Keap1 proteins, thereby inhibiting the nuclear translocation of Nrf2 and transcriptional activity [53]. In contrast, extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) were shown to cause the release of Nrf2 from Keap1 in the cytosol, and translocation of Nrf2 into the nucleus [54]. Later, Ser215, Ser408, Ser558, Thr559, Ser577 of Nrf2 were proposed as the potential targets for MAPKs and their upstream kinases [55]. The mutation of those phosphorylation sites caused a moderate decrease in the transcriptional activity of Nrf2.
In 2007, Pi et al. proposed that Nrf2 is a substrate for phosphorylation by casein kinase 2 (CK2) although phosphorylation sites were not identified [56]. In the following year, Apopa et al. showed that CK2 phosphorylated Nrf2 at multiple sites in transcription activation domains, Neh4 and Neh5, which could be dephosphorylated by λ phosphatase [57]. Increased phosphorylation of these sites correlated with the nuclear translocation of Nrf2.
In 2012, Rada et al. showed that GSK3β can phosphorylate two Ser residues, Ser344 and Ser347 (Mouse Ser335 and Ser338) of Nrf2, which are one of two binding sites for β-TrCP [41].
In 2016, Joo et al. identified Ser558 (Ser550 in mouse) of Nrf2 as a target of adenosine monophosphate (AMP)-activated protein kinase (AMPK) [58]. AMPK activation caused nuclear accumulation of Nrf2 through phosphorylation of this Ser residue located in the canonical nuclear export signal. As AMPK can inhibit GSK3β, it was proposed that AMPK can directly phosphorylate Ser550 residue, and indirectly induce dephosphorylation of Tyr568 through inhibition of the GSK3β/Fyn pathway, resulting in the nuclear accumulation of Nrf2 for the ARE-driven gene transactivation.
As discussed above, the phosphorylation of Nrf2 at the Ser40 or a neighboring region by PKC, PERK, and CK2 can enhance nuclear translocation of Nrf2 and the transcription activity. AMPK-mediated phosphorylation of Ser558 also leads to Nrf2 activation. On the other hand, the phosphorylation of Tyr576, Ser344, Ser347, or near sites by Fyn kinase, GSK3β, and other enzymes can promote the nuclear export of Nrf2, and its sequestration by Keap1 or β-TrCP, ubiquitylation, and proteasomal degradation in the cytosol, which results in the reduced transcription activity. In addition, the PI3K/PKB (Akt) pathway and AMPK can indirectly promote the nuclear translocation and transcription activity of Nrf2 by inhibiting the GSK3β-mediated pathway. Of the MAPKs, p38 appears to moderately enhance nuclear export whereas ERK and JNK enhance nuclear import of Nrf2, although their target sites are not well-defined.
5. Acetylation of Nrf2
In 2009, Sun et al. first reported the acetylation of Nrf2 as a modulatory mechanism for ARE-dependent antioxidant response. They showed that a transcriptional coactivator, p300/CBP, acetylated multiple Lys residues (Lys 438, Lys 443, Lys 445; Lys 533, Lys 536, Lys 538) within the Neh1 DNA binding domain of Nrf2, which augmented binding of Nrf2 to ARE promoter of DNA.
A regulatory role of acetylation and deacetylation of Nrf2 for transcriptional activity was further shown by Kawai et al. who verified that CBP-mediated acetylation of Nrf2 increased the binding of Nrf2 to ARE in a gene promoter, and increased the transcription of target genes [59]. They also showed that sirtuin 1 (Sirt1, a class III HDAC) could induce deacetylation of Nrf2, using a molecular approach with the expression of heterologous Sirt1 and a dominant-negative Sirt1-H355A mutant, and a pharmacological approach with the Sirt1 inhibitors, EX-527 and nicotinamide, and a putative Sirt1 activator, resveratrol. The acetylation sites were identified to be Lys596 (Mouse Lys588) and Lys599 (Mouse Lys591) of Nrf2, and the acetylation increased nuclear localization of Nrf2, whereas deacetylation enhanced its cytoplasmic localization.
In 2017, Yang et al. showed that Sirt2, a cytoplasmic sirtuin (class III HDAC) physically interacts with Nrf2 and deacetylates the Lys 506 and Lys508 residues, leading to a reduced level of nuclear Nrf2 protein, reduced ferroportin 1 (FPN1) expression, and decreased cellular iron export [60]. They further showed that Sirt2 deletion decreases cell viability under an iron deficiency condition. Moreover, livers from Sirt2(-/-) mice had increased protein levels of acetylated Nrf2 and FPN1, and decreased levels of iron, while these effects were reversed in Sirt2(-/-)/Nrf2(-/-) double knockout mice.
The acetylation of Nrf2 mainly occurred at Lys residues in the Neh1 and Neh3 domains where NES and nuclear localization signal (NLS) are located. Therefore, acetylation of Nrf2 at these regions is considered to play a regulatory role in the trafficking of Nrf2, enhancing its movement from the cytosol to the nucleus, in the opposite direction regulated by phosphorylation of Nrf2 at the Tyr576 (Mouse Tyr568) residue.
In a recent study by Ganner et al., p300 was shown to physically interact with Nrf2 and interfere with Nrf2-Keap1 complex formation [61]. The increase in acetyltransferase activity of p300 increased Nrf2 protein abundance and promoted Nrf2 nuclear localization. They proposed “a model whereby p300-mediated acetylation of Nrf2 causes dissociation of Nrf2 from Keap1, allowing Nrf2 to translocate to the nucleus and upregulate transcription of target genes, and eventually increasing survival under oxidative stress”.
6. Integrated Regulation of Nrf2 by Post-Translational Modifications and Protein–Protein Interactions
Current understanding of the regulation of Nrf2 by phosphorylation and acetylation is illustrated in Figure 4. In Figure 5, a simplified model for coordinated regulation of Nrf2 by its post-translational modification and its interaction with Keap1 or β-TrCP is schematically illustrated. It is apparently complicated, and it will become even more complicated if other known and currently unknown contributors to the flux of Nrf2 are all included [62].
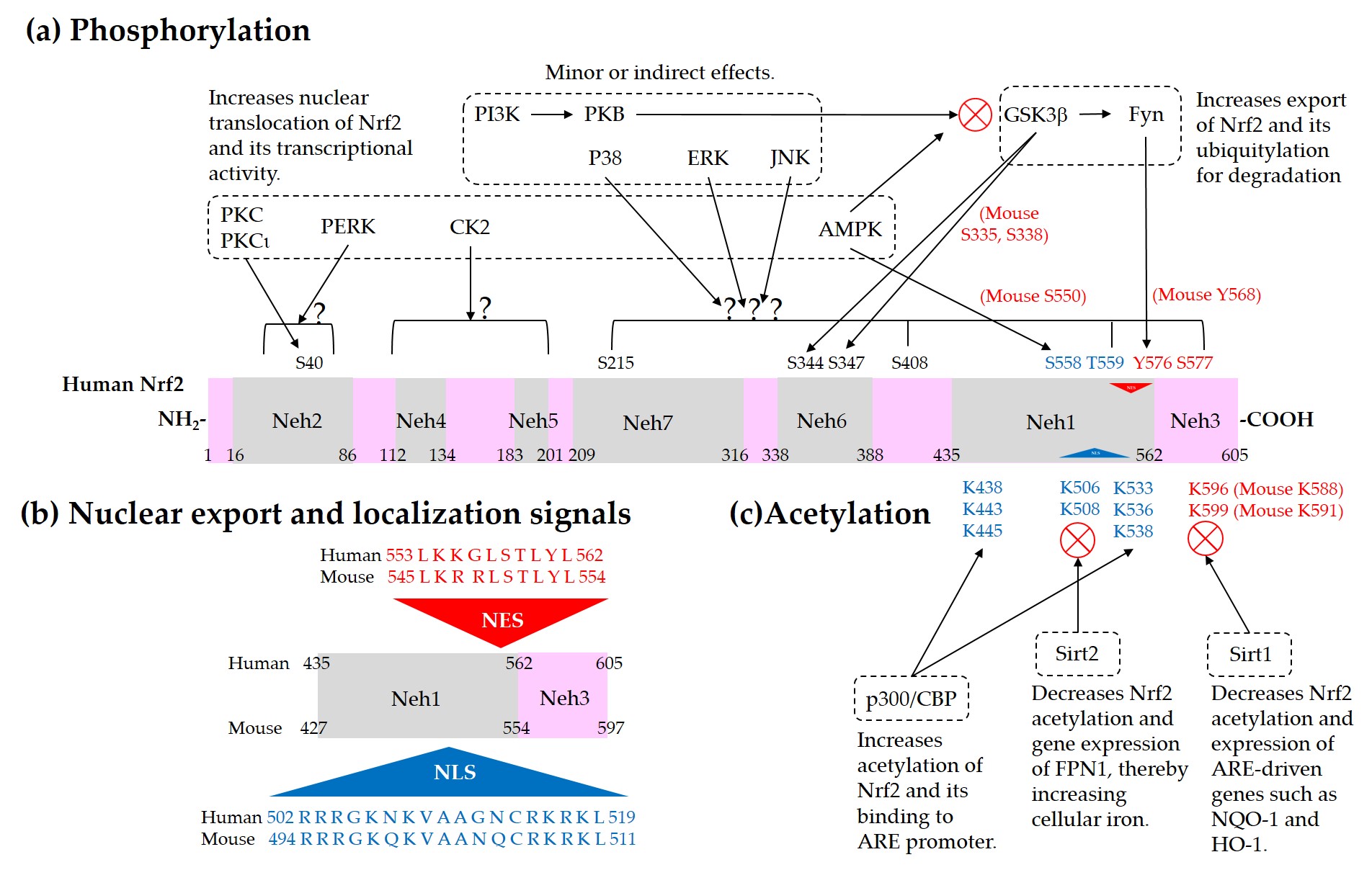
Figure 4. Regulation of Nrf2 through post-translational modifications. (a) Phosphorylation of Nrf2. Phosphorylation of Nrf2 at serine (Ser, S) 40 residue by protein kinase C (PKC) or atypical PKC-iota (ι), at Ser558 (Mouse Ser550) residue by AMP-activated protein kinase (AMPK), and at a N-terminal region by PKR-like ER kinase (PERK) can enhance nuclear translocation of Nrf2 and its transcription activity. On the other hand, phosphorylation of tyrosine (Tyr, Y) 576 (Mouse Tyr568) by a glycogen synthase kinase (GSK)3β/Fyn kinase pathway can promote the nuclear export of Nrf2 and its sequestration by Keap1, which results in the reduced transcription activity of Nrf2. The phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB, Akt) pathway and AMPK can indirectly promote the transcription activity of Nrf2 by inhibition of GSK3β activity. GSK3β can directly phosphorylate Ser344 and Ser347 (Mouse Ser335 and Ser338) in the 343DSGIS347 motif of Nrf2, enhance the binding to β-TrCP, and thus increase ubiquitylation and proteasomal degradation of Nrf2. Mitogen-activated protein kinases (MAPKs), such as p38, extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK), can phosphorylate Ser or threonine (Thr, T) residues of Nrf2 directly or indirectly, and modulate Nrf2 activity either negatively or positively, and only moderately in both cases. (b) Nuclear export signal (NES) and nuclear localization signal (NLS). Human and mouse sequences are aligned for comparison. (c) Acetylation of Nrf2. Acetylation of lysine (Lys, K) residues in the NES or NLS of Nrf2 resulting from increased levels of p300/CBP or decreased level of Sirt1 and Sirt2 can prevent nuclear export and ubiquitylation-mediated degradation of Nrf2, and increase nuclear accumulation and transcriptional activity of Nrf2.
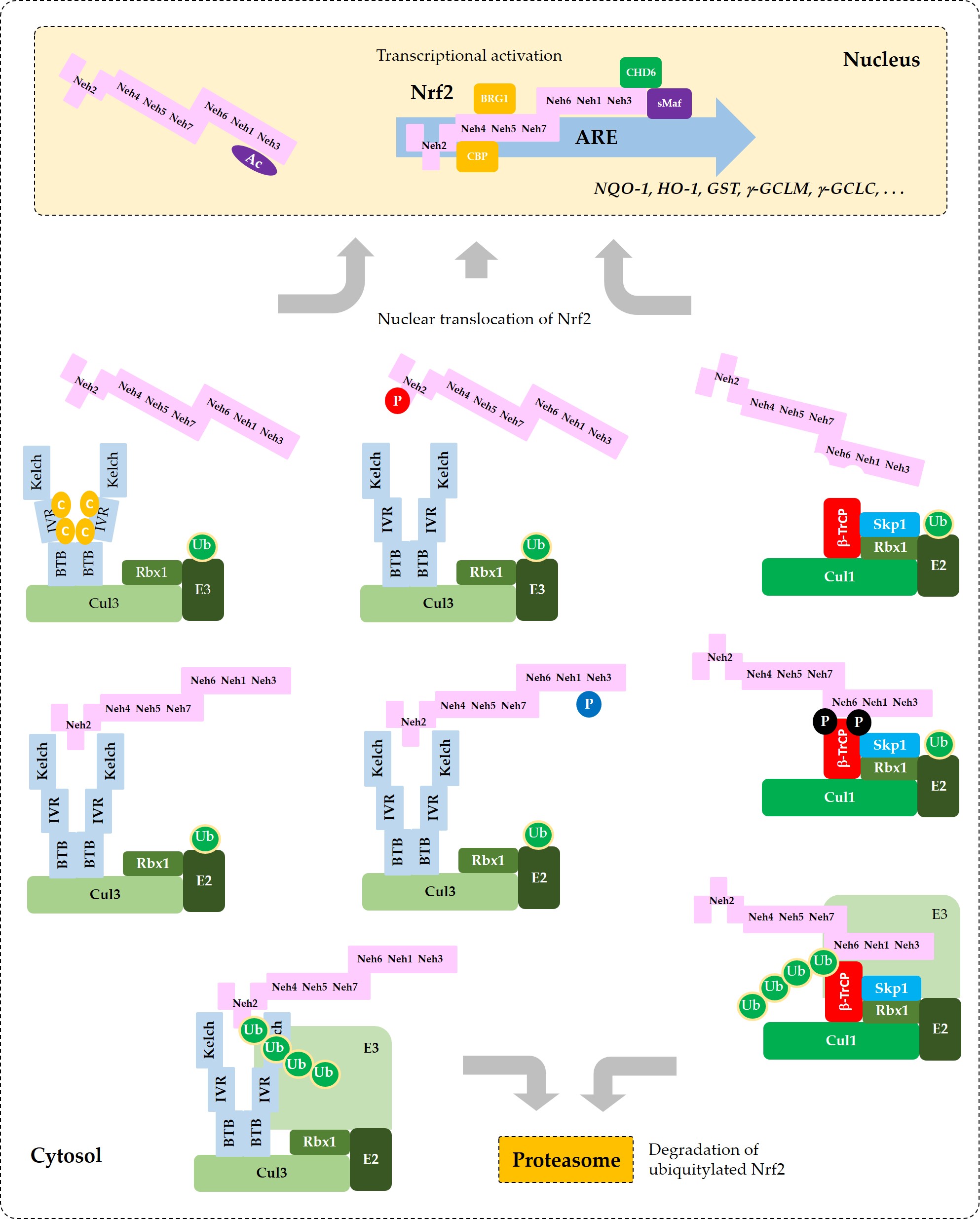
Figure 5. Coordinated regulation of Nrf2 by its post-translation modification in conjunction with its sequestration by Keap1 or β-TrCP. This schematic represents a “simplified” model for multiple states of Nrf2 that interact with Keap1 or β-TrCP with different binding affinities. Phosphorylation of Nrf2 at Tyr576 (P in blue circle) by Fyn kinase can enhance the interaction between Nrf2 and Keap1, which results in increased ubiquitylation (Ub in green circles) and subsequent proteasomal degradation of Nrf2. Phosphorylation of Nrf2 at Ser40 (P in red circle) by PKC, or the oxidative and/or chemical modification of Keap1 at Cys residues (C in yellow circles) in the IVR can weaken the interaction between Nrf2 and Keap1, which leads to an increase of Nrf2 that is released from Keap1 and enters the nucleus to transcriptionally activate ARE-driven gene expression. Phosphorylation of Nrf2 at Ser344 and Ser347 (P in black circles) by GSK3β enhances the interaction between Nrf2 and β-TrCP, which results in increased ubiquitylation and subsequent proteasomal degradation of Nrf2. If GSK3β is inhibited by PKB (Akt) or other enzymes, these Ser residues are dephosphorylated and Nrf2 can be released from β-TrCP, which allows nuclear translocation of Nrf2 and transcriptional activation. Acetylation of Lys residues (Ac in purple oval) in the NES of Nrf2 by CBP/p300 can enhance nuclear import and accumulation of Nrf2.
References
- Telakowski-Hopkins, C.A.; King, R.G.; Pickett, C.B. Glutathione S-transferase Ya subunit gene: identification of regulatory elements required for basal level and inducible expression. Proc Natl Acad Sci U S A 1988, 85, 1000-1004
- Daniel, V.; Sharon, R.; Bensimon, A. Regulatory elements controlling the basal and drug-inducible expression of glutathione S-transferase Ya subunit gene. DNA 1989, 8, 399-408
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc Natl Acad Sci U S A 1990, 87, 6258-6262
- Rushmore, T.H.; King, R.G.; Paulson, K.E.; Pickett, C.B. Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci U S A 1990, 87, 3826-3830
- Rushmore, T.H.; Pickett, C.B. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem 1990, 265, 14648-14653
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem 1991, 266, 11632-11639
- Friling, R.S.; Bergelson, S.; Daniel, V. Two adjacent AP-1-like binding sites form the electrophile-responsive element of the murine glutathione S-transferase Ya subunit gene. Proc Natl Acad Sci U S A 1992, 89, 668-672
- Orkin, S.H. Globin gene regulation and switching: circa 1990. Cell 1990, 63, 665-672
- Chan, J.Y.; Han, X.L.; Kan, Y.W. Cloning of Nrf1, an NF-E2-related transcription factor, by genetic selection in yeast. Proc Natl Acad Sci U S A 1993, 90, 11371-11375
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of Nf-E2-Related Factor-2 (Nrf2), a Nf-E2-Like Basic Leucine-Zipper Transcriptional Activator That Binds to the Tandem Nf-E2/Ap1 Repeat of the Beta-Globin Locus-Control Region. Proceedings of the National Academy of Sciences of the United States of America 1994, 91, 9926-9930
- Zhang, Y.; Qiu, L.; Li, S.; Xiang, Y.; Chen, J.; Ren, Y. The C-terminal domain of Nrf1 negatively regulates the full-length CNC-bZIP factor and its shorter isoform LCR-F1/Nrf1β; both are also inhibited by the small dominant-negative Nrf1γ/δ isoforms that down-regulate ARE-battery gene expression. PLoS One 2014, 9, e109159
- Chan, K.M.; Lu, R.H.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proceedings of the National Academy of Sciences of the United States of America 1996, 93, 13943-13948
- Tsai, J.J.; Dudakov, J.A.; Takahashi, K.; Shieh, J.H.; Velardi, E.; Holland, A.M.; Singer, N.V.; West, M.L.; Smith, O.M.; Young, L.F.; Shono, Y.; Ghosh, A.; Hanash, A.M.; Tran, H.T.; Moore, M.A.; van den Brink, M.R. Nrf2 regulates haematopoietic stem cell function. Nat Cell Biol 2013, 15, 309-316
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; Yamamoto, M.; Nabeshima, Y. An Nrf2 small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and Biophysical Research Communications 1997, 236, 313-322
- Chan, J.Y.; Kwong, M.; Lu, R.; Chang, J.; Wang, B.; Yen, T.S.; Kan, Y.W. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J 1998, 17, 1779-1787
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem 2008, 283, 33554-33562
- Kobayashi, A.; Ito, E.; Toki, T.; Kogame, K.; Takahashi, S.; Igarashi, K.; Hayashi, N.; Yamamoto, M. Molecular cloning and functional characterization of a new Cap'n' collar family transcription factor Nrf3. J Biol Chem 1999, 274, 6443-6452
- Derjuga, A.; Gourley, T.S.; Holm, T.M.; Heng, H.H.Q.; Shivdasani, R.A.; Ahmed, R.; Andrews, N.C.; Blank, V. Complexity of CNC transcription factors as revealed by gene targeting of the Nrf3 locus. Molecular and Cellular Biology 2004, 24, 3286-3294
- Liu, P.; Kerins, M.J.; Tian, W.; Neupane, D.; Zhang, D.D.; Ooi, A. Differential and overlapping targets of the transcriptional regulators NRF1, NRF2, and NRF3 in human cells. J Biol Chem 2019, 294, 18131-18149
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes & Development 1999, 13, 76-86
- Xue, F.Y.; Cooley, L. Kelch Encodes a Component of Intercellular Bridges in Drosophila Egg Chambers. Cell 1993, 72, 681-693
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and Characterization of a Novel Erythroid Cell-Derived Cnc Family Transcription Factor Heterodimerizing with the Small Maf Family Proteins. Molecular and Cellular Biology 1995, 15, 4184-4193
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O'Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes to Cells 2003, 8, 379-391
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. Journal of Biological Chemistry 2002, 277, 36544-36552
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proceedings of the National Academy of Sciences of the United States of America 2002, 99, 11908-11913
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Molecular and Cellular Biology 2003, 23, 8137-8151
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Molecular and Cellular Biology 2004, 24, 10941-10953
- Kobayashi, M.; Itoh, K.; Suzuki, T.; Osanai, H.; Nishikawa, K.; Katoh, Y.; Takagi, Y.; Yamamoto, M. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells 2002, 7, 807-820
- Katoh, Y.; Iida, K.; Kang, M.I.; Kobayashi, A.; Mizukami, M.; Tong, K.I.; McMahon, M.; Hayes, J.D.; Itoh, K.; Yamamoto, M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Archives of Biochemistry and Biophysics 2005, 433, 342-350
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Molecular and Cellular Biology 2006, 26, 2887-2900
- Cullinan, S.B.; Gordan, J.D.; Jin, J.O.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Molecular and Cellular Biology 2004, 24, 8477-8486
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate for proteasomal degradation of Nrf2. Molecular and Cellular Biology 2004, 24, 7130-7139
- Furukawa, M.; Xiong, Y. BTB protein keap1 targets antioxidant transcription factor nrf2 for ubiquitination by the cullin 3-Roc1 ligase. Molecular and Cellular Biology 2005, 25, 162-171
- Velichkova, M.; Hasson, T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Molecular and Cellular Biology 2005, 25, 4501-4513
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568-572
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857-868
- Zhang, J.; Hosoya, T.; Maruyama, A.; Nishikawa, K.; Maher, J.M.; Ohta, T.; Motohashi, H.; Fukamizu, A.; Shibahara, S.; Itoh, K.; Yamamoto, M. Nrf2 Neh5 domain is differentially utilized in the transactivation of cytoprotective genes. Biochem J 2007, 404, 459-466
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H : quinone oxidoreductase 1 gene expression and induction in response to antioxidants. Journal of Biological Chemistry 2005, 280, 16891-16900
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol 2005, 25, 10895-10906
- Rojo, A.I.; Medina-Campos, O.N.; Rada, P.; Zuniga-Toala, A.; Lopez-Gazcon, A.; Espada, S.; Pedraza-Chaverri, J.; Cuadrado, A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. Free Radic Biol Med 2012, 52, 473-487
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; Hayes, J.D.; Bertho, G.; Cuadrado, A. Structural and Functional Characterization of Nrf2 Degradation by the Glycogen Synthase Kinase 3/beta-TrCP Axis. Molecular and Cellular Biology 2012, 32, 3486-3499
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765-3781
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radical Biology and Medicine 2015, 88, 147-157
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev 2014, 28, 708-722
- Li, W.; Liu, H.; Zhou, J.S.; Cao, J.F.; Zhou, X.B.; Choi, A.M.K.; Chen, Z.H.; Shen, H.H. Caveolin-1 Inhibits Expression of Antioxidant Enzymes through Direct Interaction with Nuclear Erythroid 2 p45-related Factor-2 (Nrf2). Journal of Biological Chemistry 2012, 287, 20922-20930
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. Journal of Biological Chemistry 2002, 277, 42769-42774
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element - Degradation of Nrf2 by the 26 S proteasome. Journal of Biological Chemistry 2003, 278, 4536-4541
- Numazawa, S.; Ishikawa, M.; Yoshida, A.; Tanaka, S.; Yoshida, T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. American Journal of Physiology-Cell Physiology 2003, 285, C334-C342
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Molecular and Cellular Biology 2003, 23, 7198-7209
- Jain, A.K.; Jaiswal, A.K. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J Biol Chem 2006, 281, 12132-12142
- Jain, A.K.; Jaiswal, A.K. GSK-3 beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. Journal of Biological Chemistry 2007, 282, 16502-16510
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem 2006, 281, 14841-14851
- Keum, Y.S.; Yu, S.W.; Chang, P.P.J.; Yuan, X.L.; Kim, J.H.; Xu, C.J.; Han, J.H.; Agarwal, A.; Kong, A.N.T. Mechanism of action of sulforaphane: Inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Research 2006, 66, 8804-8813
- Yuan, X.L.; Xu, C.J.; Pan, Z.; Keum, Y.S.; Kim, J.H.; Shen, G.X.; Yu, S.W.; Oo, K.T.; Ma, J.J.; Kong, A.N.T. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Molecular Carcinogenesis 2006, 45, 841-850
- Sun, Z.; Huang, Z.P.; Zhang, D.D. Phosphorylation of Nrf2 at Multiple Sites by MAP Kinases Has a Limited Contribution in Modulating the Nrf2-Dependent Antioxidant Response. Plos One 2009, 4, e6588
- Pi, J.B.; Bai, Y.S.; Reece, J.M.; Williams, J.; Liu, D.X.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; Collins, S.; Waalkes, M.P. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radical Biology and Medicine 2007, 42, 1797-1806
- Apopa, P.L.; He, X.Q.; Ma, Q. Phosphorylation of nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. Journal of Biochemical and Molecular Toxicology 2008, 22, 63-76
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Molecular and Cellular Biology 2016, 36, 1931-1942
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.Q.; Arinze, I.J. Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization. Journal of Biological Chemistry 2011, 286, 7629-7640
- Yang, X.; Park, S.H.; Chang, H.C.; Shapiro, J.S.; Vassilopoulos, A.; Sawicki, K.T.; Chen, C.; Shang, M.; Burridge, P.W.; Epting, C.L.; Wilsbacher, L.D.; Jenkitkasemwong, S.; Knutson, M.; Gius, D.; Ardehali, H. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2. J Clin Invest 2017, 127, 1505-1516
- Ganner, A.; Pfeiffer, Z.C.; Wingendorf, L.; Kreis, S.; Klein, M.; Walz, G.; Neumann-Haefelin, E. The acetyltransferase p300 regulates NRF2 stability and localization. Biochemical and Biophysical Research Communications 2020, 524, 895-902
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol 2018, 17, 297-314