The highly vascularized URT is the primary route of ingress of inhaled pathogens. A dense network of mucosal-associated lymphoid tissues (MALTs) is in the mucosal tissues to help induce pathogen-specific immune responses, reducing occurrences of infections.
- DNA vaccine
- nanoparticles
- mucosal vaccine
- pulmonary delivery
1. Background
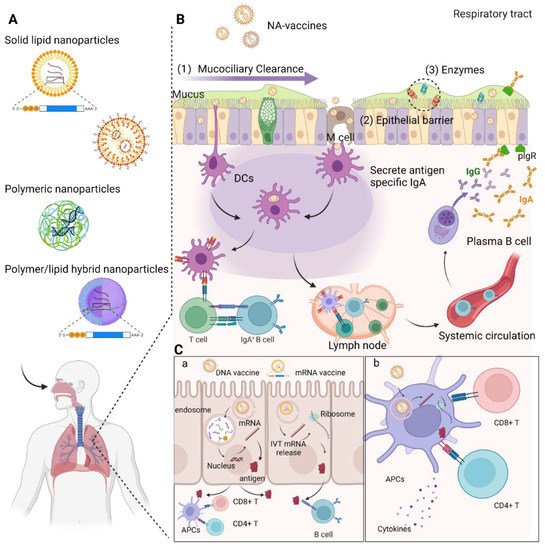
Recent advancements in the field of in vitro transcribed mRNA (IVT-mRNA) vaccination have attracted considerable attention to such vaccination as a cutting-edge technique against infectious diseases including COVID-19 caused by SARS-CoV-2. While numerous pathogens infect the host through the respiratory mucosa, conventional parenterally administered vaccines are unable to induce protective immunity at mucosal surfaces. Mucosal immunization enables the induction of both mucosal and systemic immunity, efficiently removing pathogens from the mucosa before an infection occurs. Although respiratory mucosal vaccination is highly appealing, successful nasal or pulmonary delivery of nucleic acid-based vaccines is challenging because of several physical and biological barriers at the airway mucosal site, such as a variety of protective enzymes and mucociliary clearance, which remove exogenously inhaled substances. Hence, advanced nanotechnologies enabling delivery of DNA and IVT-mRNA to the nasal and pulmonary mucosa are urgently needed.
2. DNA Vaccines
2.1. Delivery of DNA Vaccines via Respiratory Routes
2.2. Delivery Systems for DNA Vaccines via Respiratory Routes
2.2.1. Liposomes and Niosomes
2.2.2. Polymers
Polyethylenimine
Chitosan
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468.
- Ulmer, J.B.; Donnelly, J.J.; Parker, S.E.; Rhodes, G.H.; Felgner, P.L.; Dwarki, V.J.; Gromkowski, S.H.; Deck, R.R.; DeWitt, C.M.; Friedman, A.; et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science 1993, 259, 1745–1749.
- Silveira, M.M.; Moreira, G.M.S.G.; Mendonça, M. DNA vaccines against COVID-19: Perspectives and challenges. Life Sci. 2021, 267, 118919.
- Ingolotti, M.; Kawalekar, O.; Shedlock, D.J.; Muthumani, K.; Weiner, D.B. DNA vaccines for targeting bacterial infections. Expert Rev. Vaccines 2010, 9, 747–763.
- Xu, Y.; Yuen, P.-W.; Lam, J.K.-W. Intranasal DNA Vaccine for Protection against Respiratory Infectious Diseases: The Delivery Perspectives. Pharmaceutics 2014, 6, 378–415.
- Mallapaty, S.; Callaway, E. What scientists do and don’t know about the Oxford-AstraZeneca COVID vaccine. Nature 2021, 592, 15–17.
- Kutzler, M.A.; Weiner, D.B. DNA vaccines: Ready for prime time? Nat. Rev. Genet. 2008, 9, 776–788.
- Tebas, P.; Yang, S.; Boyer, J.D.; Reuschel, E.L.; Patel, A.; Christensen-Quick, A.; Andrade, V.M.; Morrow, M.P.; Kraynyak, K.; Agnes, J.; et al. Safety and immunogenicity of INO-4800 DNA vaccine against SARS-CoV-2: A preliminary report of an open-label, Phase 1 clinical trial. EClinicalMedicine 2020, 31, 100689.
- Modjarrad, K.; Roberts, C.C.; Mills, K.T.; Castellano, A.R.; Paolino, K.; Muthumani, K.; Reuschel, E.L.; Robb, M.L.; Racine, T.; Oh, M.-D.; et al. Safety and immunogenicity of an anti-Middle East respiratory syndrome coronavirus DNA vaccine: A phase 1, open-label, single-arm, dose-escalation trial. Lancet. Infect. Dis. 2019, 19, 1013–1022.
- Sheridan, C. First COVID-19 DNA vaccine approved, others in hot pursuit. Nat. Biotechnol. 2021, 39, 1479–1482.
- Mallapaty, S. India’s DNA COVID vaccine is a world first—More are coming. Nature 2021, 597, 161–162.
- Lombry, C.; Marteleur, A.; Arras, M.; Lison, D.; Louahed, J.; Renauld, J.-C.; Préat, V.; Vanbever, R. Local and systemic immune responses to intratracheal instillation of antigen and DNA vaccines in mice. Pharm. Res. 2004, 21, 127–135.
- Bivas-Benita, M.; van Meijgaarden, K.E.; Franken, K.L.M.C.; Junginger, H.E.; Borchard, G.; Ottenhoff, T.H.M.; Geluk, A. Pulmonary delivery of chitosan-DNA nanoparticles enhances the immunogenicity of a DNA vaccine encoding HLA-A*0201-restricted T-cell epitopes of Mycobacterium tuberculosis. Vaccine 2004, 22, 1609–1615.
- Low, L.; Mander, A.; McCann, K.; Dearnaley, D.; Tjelle, T.; Mathiesen, I.; Stevenson, F.; Ottensmeier, C.H. DNA vaccination with electroporation induces increased antibody responses in patients with prostate cancer. Hum. Gene Ther. 2009, 20, 1269–1278.
- Rottinghaus, S.T.; Poland, G.A.; Jacobson, R.M.; Barr, L.J.; Roy, M.J. Hepatitis B DNA vaccine induces protective antibody responses in human non-responders to conventional vaccination. Vaccine 2003, 21, 4604–4608.
- Brito, L.A.; Malyala, P.; O’Hagan, D.T. Vaccine adjuvant formulations: A pharmaceutical perspective. Semin. Immunol. 2013, 25, 130–145.
- Legendre, J.Y.; Szoka, F.C.J. Delivery of plasmid DNA into mammalian cell lines using pH-sensitive liposomes: Comparison with cationic liposomes. Pharm. Res. 1992, 9, 1235–1242.
- Khatri, K.; Goyal, A.K.; Gupta, P.N.; Mishra, N.; Mehta, A.; Vyas, S.P. Surface modified liposomes for nasal delivery of DNA vaccine. Vaccine 2008, 26, 2225–2233.
- Gogev, S.; de Fays, K.; Versali, M.-F.; Gautier, S.; Thiry, E. Glycol chitosan improves the efficacy of intranasally administrated replication defective human adenovirus type 5 expressing glycoprotein D of bovine herpesvirus 1. Vaccine 2004, 22, 1946–1953.
- Lay, M.; Callejo, B.; Chang, S.; Hong, D.K.; Lewis, D.B.; Carroll, T.D.; Matzinger, S.; Fritts, L.; Miller, C.J.; Warner, J.F.; et al. Cationic lipid/DNA complexes (JVRS-100) combined with influenza vaccine (Fluzone) increases antibody response, cellular immunity, and antigenically drifted protection. Vaccine 2009, 27, 3811–3820.
- D’Souza, S.; Rosseels, V.; Denis, O.; Tanghe, A.; De Smet, N.; Jurion, F.; Palfliet, K.; Castiglioni, N.; Vanonckelen, A.; Wheeler, C.; et al. Improved tuberculosis DNA vaccines by formulation in cationic lipids. Infect. Immun. 2002, 70, 3681–3688.
- Rosada, R.S.; de la Torre, L.G.; Frantz, F.G.; Trombone, A.P.F.; Zárate-Bladés, C.R.; Fonseca, D.M.; Souza, P.R.M.; Brandão, I.T.; Masson, A.P.; Soares, E.G.; et al. Protection against tuberculosis by a single intranasal administration of DNA-hsp65 vaccine complexed with cationic liposomes. BMC Immunol. 2008, 9, 38.
- Wong, J.P.; Zabielski, M.A.; Schmaltz, F.L.; Brownlee, G.G.; Bussey, L.A.; Marshall, K.; Borralho, T.; Nagata, L.P. DNA vaccination against respiratory influenza virus infection. Vaccine 2001, 19, 2461–2467.
- Wang, D.; Christopher, M.E.; Nagata, L.P.; Zabielski, M.A.; Li, H.; Wong, J.P.; Samuel, J. Intranasal immunization with liposome-encapsulated plasmid DNA encoding influenza virus hemagglutinin elicits mucosal, cellular and humoral immune responses. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2004, 31 (Suppl. 1), 99–106.
- Chen, C.; Han, D.; Cai, C.; Tang, X. An overview of liposome lyophilization and its future potential. J. Control. Release 2010, 142, 299–311.
- Mishima, K. Biodegradable particle formation for drug and gene delivery using supercritical fluid and dense gas. Adv. Drug Deliv. Rev. 2008, 60, 411–432.
- Lo, Y.; Tsai, J.; Kuo, J. Liposomes and disaccharides as carriers in spray-dried powder formulations of superoxide dismutase. J. Control. Release 2004, 94, 259–272.
- Kadimi, U.S.; Balasubramanian, D.R.; Ganni, U.R.; Balaraman, M.; Govindarajulu, V. In vitro studies on liposomal amphotericin B obtained by supercritical carbon dioxide-mediated process. Nanomedicine 2007, 3, 273–280.
- Baldino, L.; Reverchon, E. Niosomes formation using a continuous supercritical CO2 assisted process. J. CO2 Util. 2021, 52, 101669.
- Durak, S.; Esmaeili Rad, M.; Alp Yetisgin, A.; Eda Sutova, H.; Kutlu, O.; Cetinel, S.; Zarrabi, A. Niosomal Drug Delivery Systems for Ocular Disease-Recent Advances and Future Prospects. Nanomaterials 2020, 10, 1191.
- Grijalvo, S.; Puras, G.; Zárate, J.; Sainz-Ramos, M.; Qtaish, N.A.L.; López, T.; Mashal, M.; Attia, N.; Díaz, D.; Pons, R.; et al. Cationic Niosomes as Non-Viral Vehicles for Nucleic Acids: Challenges and Opportunities in Gene Delivery. Pharmaceutics 2019, 11, 50.
- Rose, J.K.; Buonocore, L.; Whitt, M.A. A new cationic liposome reagent mediating nearly quantitative transfection of animal cells. Biotechniques 1991, 10, 520–525.
- Manosroi, J.; Khositsuntiwong, N.; Manosroi, W.; Götz, F.; Werner, R.G.; Manosroi, A. Enhancement of transdermal absorption, gene expression and stability of tyrosinase plasmid (pMEL34)-loaded elastic cationic niosomes: Potential application in vitiligo treatment. J. Pharm. Sci. 2010, 99, 3533–3541.
- Perrie, Y.; Barralet, J.E.; McNeil, S.; Vangala, A. Surfactant vesicle-mediated delivery of DNA vaccines via the subcutaneous route. Int. J. Pharm. 2004, 284, 31–41.
- Jain, S.; Singh, P.; Mishra, V.; Vyas, S.P. Mannosylated niosomes as adjuvant-carrier system for oral genetic immunization against hepatitis B. Immunol. Lett. 2005, 101, 41–49.
- Singh, M.; Chakrapani, A.; O’Hagan, D. Nanoparticles and microparticles as vaccine-delivery systems. Expert Rev. Vaccines 2007, 6, 797–808.
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.C.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154.
- Gratton, S.E.A.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618.
- Blank, F.; Wehrli, M.; Lehmann, A.; Baum, O.; Gehr, P.; von Garnier, C.; Rothen-Rutishauser, B.M. Macrophages and dendritic cells express tight junction proteins and exchange particles in an in vitro model of the human airway wall. Immunobiology 2011, 216, 86–95.
- Thiele, L.; Rothen-Rutishauser, B.; Jilek, S.; Wunderli-Allenspach, H.; Merkle, H.P.; Walter, E. Evaluation of particle uptake in human blood monocyte-derived cells in vitro. Does phagocytosis activity of dendritic cells measure up with macrophages? J. Control. Release 2001, 76, 59–71.
- De Temmerman, M.-L.; Rejman, J.; Demeester, J.; Irvine, D.J.; Gander, B.; De Smedt, S.C. Particulate vaccines: On the quest for optimal delivery and immune response. Drug Discov. Today 2011, 16, 569–582.
- Boussif, O.; LezoualC’H, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301.
- Densmore, C.L.; Orson, F.M.; Xu, B.; Kinsey, B.M.; Waldrep, J.C.; Hua, P.; Bhogal, B.; Knight, V. Aerosol Delivery of Robust Polyethyleneimine-DNA Complexes for Gene Therapy and Genetic Immunization. Mol. Ther. 2000, 1, 180–188.
- Gautam, A.; Densmore, C.L.; Xu, B.; Waldrep, J.C. Enhanced gene expression in mouse lung after PEI-DNA aerosol delivery. Mol. Ther. 2000, 2, 63–70.
- Shim, B.-S.; Park, S.-M.; Quan, J.-S.; Jere, D.; Chu, H.; Song, M.K.; Kim, D.W.; Jang, Y.-S.; Yang, M.-S.; Han, S.H.; et al. Intranasal immunization with plasmid DNA encoding spike protein of SARS-coronavirus/polyethylenimine nanoparticles elicits antigen-specific humoral and cellular immune responses. BMC Immunol. 2010, 11, 65.
- Torrieri-Dramard, L.; Lambrecht, B.; Ferreira, H.L.; Van den Berg, T.; Klatzmann, D.; Bellier, B. Intranasal DNA vaccination induces potent mucosal and systemic immune responses and cross-protective immunity against influenza viruses. Mol. Ther. 2011, 19, 602–611.
- Bivas-Benita, M.; Bar, L.; Gillard, G.O.; Kaufman, D.R.; Simmons, N.L.; Hovav, A.-H.; Letvin, N.L. Efficient Generation of Mucosal and Systemic Antigen-Specific CD8+ T-Cell Responses following Pulmonary DNA Immunization. J. Virol. 2010, 84, 5764–5774.
- Bivas-Benita, M.; Gillard, G.O.; Bar, L.; White, K.A.; Webby, R.J.; Hovav, A.H.; Letvin, N.L. Airway CD8+ T cells induced by pulmonary DNA immunization mediate protective anti-viral immunity. Mucosal Immunol. 2013, 6, 156–166.
- Regnström, K.; Ragnarsson, E.G.E.; Köping-Höggård, M.; Torstensson, E.; Nyblom, H.; Artursson, P. PEI—A potent, but not harmless, mucosal immuno-stimulator of mixed T-helper cell response and FasL-mediated cell death in mice. Gene Ther. 2003, 10, 1575–1583.
- Mann, J.F.S.; McKay, P.F.; Arokiasamy, S.; Patel, R.K.; Klein, K.; Shattock, R.J. Pulmonary delivery of DNA vaccine constructs using deacylated PEI elicits immune responses and protects against viral challenge infection. J. Control. Release 2013, 170, 452–459.
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347.
- Bivas-Benita, M.; Romeijn, S.; Junginger, H.E.; Borchard, G. PLGA-PEI nanoparticles for gene delivery to pulmonary epithelium. Eur. J. Pharm. Biopharm. Off. J. Arb. fur Pharm. Verfahr. e.V 2004, 58, 1–6.
- Oster, C.G.; Kim, N.; Grode, L.; Barbu-Tudoran, L.; Schaper, A.K.; Kaufmann, S.H.E.; Kissel, T. Cationic microparticles consisting of poly(lactide-co-glycolide) and polyethylenimine as carriers systems for parental DNA vaccination. J. Control. Release 2005, 104, 359–377.
- Kang, M.L.; Cho, C.S.; Yoo, H.S. Application of chitosan microspheres for nasal delivery of vaccines. Biotechnol. Adv. 2009, 27, 857–865.
- Casettari, L.; Vllasaliu, D.; Lam, J.K.W.; Soliman, M.; Illum, L. Biomedical applications of amino acid-modified chitosans: A review. Biomaterials 2012, 33, 7565–7583.
- Issa, M.M.; Köping-Höggård, M.; Artursson, P. Chitosan and the mucosal delivery of biotechnology drugs. Drug Discov. Today. Technol. 2005, 2, 1–6.
- Seferian, P.G.; Martinez, M.L. Immune stimulating activity of two new chitosan containing adjuvant formulations. Vaccine 2000, 19, 661–668.
- Kumar, M.; Behera, A.K.; Lockey, R.F.; Zhang, J.; Bhullar, G.; De La Cruz, C.P.; Chen, L.-C.; Leong, K.W.; Huang, S.-K.; Mohapatra, S.S. Intranasal gene transfer by chitosan-DNA nanospheres protects BALB/c mice against acute respiratory syncytial virus infection. Hum. Gene Ther. 2002, 13, 1415–1425.
- Iqbal, M.; Lin, W.; Jabbal-Gill, I.; Davis, S.S.; Steward, M.W.; Illum, L. Nasal delivery of chitosan-DNA plasmid expressing epitopes of respiratory syncytial virus (RSV) induces protective CTL responses in BALB/c mice. Vaccine 2003, 21, 1478–1485.
- Raghuwanshi, D.; Mishra, V.; Das, D.; Kaur, K.; Suresh, M.R. Dendritic cell targeted chitosan nanoparticles for nasal DNA immunization against SARS-CoV nucleocapsid protein. Mol. Pharm. 2012, 9, 946–956.
- Wu, M.; Zhao, H.; Li, M.; Yue, Y.; Xiong, S.; Xu, W. Intranasal vaccination with mannosylated chitosan formulated DNA vaccine enables robust IgA and cellular response induction in the lungs of mice and improves protection against pulmonary mycobacterial challenge. Front. Cell. Infect. Microbiol. 2017, 7, 445.
- Bernkop-Schnürch, A.; Hornof, M.; Guggi, D. Thiolated chitosans. Eur. J. Pharm. Biopharm. Off. J. Arb. fur Pharm. Verfahrenstechnik e.V 2004, 57, 9–17.
- Nafee, N.; Taetz, S.; Schneider, M.; Schaefer, U.F.; Lehr, C.-M. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: Effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine 2007, 3, 173–183.
- Janes, K.A.; Calvo, P.; Alonso, M.J. Polysaccharide colloidal particles as delivery systems for macromolecules. Adv. Drug Deliv. Rev. 2001, 47, 83–97.
- Ravi Kumar, M.N.V.; Bakowsky, U.; Lehr, C.M. Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomaterials 2004, 25, 1771–1777.
- Wang, G.; Pan, L.; Zhang, Y.; Wang, Y.; Zhang, Z.; Lü, J.; Zhou, P.; Fang, Y.; Jiang, S. Intranasal delivery of cationic PLGA nano/microparticles-loaded FMDV DNA vaccine encoding IL-6 elicited protective immunity against FMDV challenge. PLoS ONE 2011, 6, e27605.