Kidney cancer, also known as renal cancer, is defined as a disease that starts in the kidneys. It is one of the top 10 most common cancers in the United States with more than 76,000 new cases diagnosed each year. A retrospective cohort study found that 26% of kidney cancer patients had CKD before tumor nephrectomy
[66][101]. The cause of developing kidney cancer is still not clear. However, there are several factors that can increase the risk of kidney cancer, such as older age, smoking, obesity, hypertension, and being on kidney dialysis. RCC, also known as renal cell adenocarcinoma, is the most common type of kidney cancer in adults. A study demonstrated the risk of RCC in ESRD patients is increased up to 100 times
[67][102]. RCC denotes cancer originated from the renal epithelium and accounts for >90% of cancers in the kidney and can be distinguished by histopathological features and gene mutations
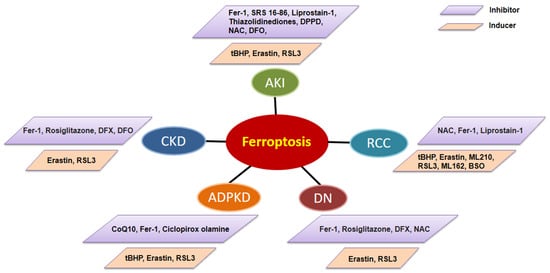
[68][103]. Localized RCC can be successfully manage with surgery, whereas metastatic RCC is refractory to conventional chemotherapy in which most patients show resistance to chemotherapy and radiotherapy. Activation of regulated cell death is considered an ideal therapeutic strategy for cancer and may help drug resistance. Research on the effect of erastin in 60 tumor cell lines of eight tissues found that RCC cells are more susceptible than others to erastin induced cell death
[69][104]. Clear cell renal cell carcinoma (ccRCC) is the most common type of RCC. Both hereditary and familial ccRCC are strongly connected with von Hippel Lindau (VHL) gene mutations, which consecutively lead to the stabilization of hypoxia-inducible transcription factor (HIF)
[70][105]. Miess et al. demonstrated that the silence of genes coding for glutathione peroxidases, GPX3 and GPX4, is lethal to ccRCC cells
[71][106]. They also found that the cell viability of ccRCC is dependent on the synthesis of GSH, which prevents the accumulation of lipid peroxides. In addition, the re-expressed VHL gene had a resistance effect on ferroptosis in VHL-deficient RCC cells
[71][106]. In addition, the susceptibility of ferroptosis can be affected by cell density and this effect is mediated by TAZ through the regulation of EMP1-NOX4 in ccRCC
[72][107]. Furthermore, a 2–82% mutation rate among 36 ferroptosis associated genes (FRGs), including TP53, NFE2L2, FANCD2, DPP4, ALOX5, PTGS2, ALOX15B, ACSL4, CARS, and HMGCR, was detected in ccRCC in an analysis based on the GSCA database
[73][108]. A new survival model was built based on five risk-related FRGs (CARS, NCOA4, FANCD2, HMGCR, and SLC7A11), which indicated that high expression of FANCD2, CARS, and SLC7A11 and low expression of HMGCR and NCOA4 are associated with high-risk ccRCC patients. These studies suggest that FRGs are the potential prognostic biomarkers and ferroptosis modulation may have therapeutic potentials in ccRCC.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) are the autosomal dominant disorder caused by germline mutations in fumarate hydratase (FH), which is characterized by multiple cutaneous piloleiomyomas, uterine leiomyomas, and papillary type 2 renal cancer, which is resistant to current radiotherapy, chemotherapy, and immunotherapy
[74][109]. It has been suggested that accumulated fumarate drives constitutive Nrf2 activation, which promotes the transcription of FTL and FTH1 genes in HLRCC cells in vitro
[75][110]. On one side, excessive fumarate inhibits IRPs’ ability via the repression of FTL and FTH1 mRNA translation, which results in high intracellular ferritin levels
[76][111]. High intracellular ferritin further sequesters free iron and finally results in a drop in the labile iron pool. On the other side, the FH accumulation sensitizes HLRCC cells to ferroptosis through C93 modification which represses GPX4 activity
[77][112]. FH is also shown to indirectly inhibit AMPK, resulting in indirect inhibition of DMT1 expression
[76][111]. This prevents the efflux of iron from the endosome into the cytoplasm to further reduce the labile iron pool. Induction of ferroptosis in FH-inactivated tumors represents an opportunity for synthetic lethality in cancer. Thus, pharmacological suppression of those proteins represents a treatment strategy worth exploring.