The oxidation of cumene and following cleavage of cumene hydroperoxide (CHP) with sulfuric acid (Hock rearrangement) is still, by far, the dominant synthetic route to produce phenol. In 2020, the global phenol market reached a value of 23.3 billion US$ with a projected compound annual growth rate of 3.4% for 2020–2025. From ecological and economical viewpoints, the key step of this process is the cleavage of CHP. One sought-after way to likewise reduce energy consumption and waste production of the process is to substitute sulfuric acid with heterogeneous catalysts. Different types of zeolites, silicon-based clays, heteropoly acids, and ion exchange resins have been investigated and tested in various studies. For every type of these solid acid catalysts, several materials were found that show high yield and selectivity to phenol.
- Hock cleavage
- heterogeneous catalysis
- cumene hydroperoxide
1. Introduction
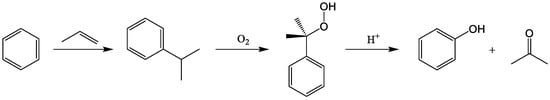
2. Solid Acid Catalysts
After the commercialization in Europe and the USA after World War II, different other homogeneous catalysts have been investigated [38[20][21][22][23][24][25][26],39,40,41,42,43,44], but none of them demonstrated superior performance to sulfuric acid regarding catalytic and cost efficiency. To eliminate the persisting issues with sulfuric acid as corrosive and homogeneous catalyst, different approaches to substitute the homogeneous catalyst with a solid catalyst have been explored in the past 20 years.2.1. Mineral Acid-Treated Clays
2.2. Heteropoly Acids (HPA) on Supports
2.3. Zeolites
2.4. Ion Exchange Resins
| Catalyst | Si/Al Ratio | Reaction Time/min | Reaction Temperature/°C | Conversion of CHP/% | Y (phenol)/% | S (phenol)/% | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clays | |||||||||||||||
| K10 | 51 | n/a | 30 | 50 | 100 | n/a | n/a | ||||||||
| K10 | 51 | with ZnCl | 2 | n/a | 30 | 50 | 80 | n/a | n/a | ||||||
| K10 | 51 | with FeCl | 3 | n/a | 30 | 50 | 92 | n/a | n/a | ||||||
| K10 | 51 | with CeCl | 3 | n/a | 30 | 50 | 100 | n/a | n/a | ||||||
| K10 | 51 | with LaCl | 3 | n/a | 30 | 50 | 95 | n/a | n/a | ||||||
| K10 | 18 | n/a | 30 | 40 | 99 | n/a | n/a | ||||||||
| Grade F24 | 52 | n/a | 120 | 57–80 | 100 | 98 | 98 | ||||||||
| Grade F113 | 52 | n/a | 120 | 57–80 | 100 | 95 | 95 | ||||||||
| Grade F13 | 52 | n/a | 120 | 57–80 | 100 | 99 | 99 | ||||||||
| Grade F62 | 52 | n/a | 120 | 57–80 | 76 | 37 | 49 | ||||||||
| Acidic montmorillonite | 53 | n/a | 90 | 90 | n/a | 95 | n/a | ||||||||
| HPA on supports | |||||||||||||||
| 12-tungstophosphoric acid | 52 | n/a | 120 | 57–80 | 100 | 99 | n/a | ||||||||
| 12-molybdophosphoric acid | 52 | n/a | 120 | 57–80 | 100 | 99 | n/a | ||||||||
| 12-tungstosilicic acid | 52 | n/a | 120 | 57–80 | 100 | 99 | n/a | ||||||||
| 12-molybdosilicic acid | 52 | n/a | 120 | 57–80 | 100 | 99 | n/a | ||||||||
| Cs | 2.5 | H | 0.5 | PW | 12 | O | 40 | on K10 | 18 | n/a | 30 | 40 | 99 | n/a | |
| Zeolites | |||||||||||||||
| H-[Al]-Beta | 65 | 14 | 5 | 25 | 100 | 88 | 88 | ||||||||
| H-[Ga]-Beta | 65 | 20 | 5 | 25 | 100 | 92 | 92 | ||||||||
| H-[Fe]-Beta | 65 | 22 | 5 | 25 | 100 | 91 | 91 | ||||||||
| H-[B]-Beta | 65 | 30 | 5 | 25 | 100 | 92 | 92 | ||||||||
| H-[Al]-ZSM-5 | 65 | 30 | 5 | 25 | 100 | 86 | 86 | ||||||||
| H-[Ga]-ZSM-5 | 65 | 35 | 5 | 25 | 100 | 89 | 89 | ||||||||
| H-[Fe]-ZSM-5 | 65 | 30 | 5 | 25 | 100 | 88 | 88 | ||||||||
| H-Mordenite | 65 | 7 | 5 | 25 | 100 | 87 | 87 | ||||||||
| H-Y | 65 | 2.5 | 10 | 40 | 96 | 82 | 85 | ||||||||
| H-[Al]-ZSM-12 | 65 | 40 | 30 | 40 | 95 | 78 | 82 | ||||||||
| H-[Al]-NCL-1 | 65 | 40 | 15 | 40 | 85 | 71 | 84 | ||||||||
| H-[Al]-ZSM-22 | 65 | 60 | 15 | 40 | 65 | 57 | 88 | ||||||||
| H-[Al]-MCM-22 | 65 | 30 | 15 | 40 | 90 | 78 | 87 | ||||||||
| H-[Al]-ZSM-48 | 65 | 50 | 60 | 60 | 45 | 36 | 80 | ||||||||
| H-[Al]-EU-1 | 65 | 50 | 30 | 60 | 80 | 71 | 89 | ||||||||
| H-SAPO-5 | 65 | n/a | 60 | 60 | 10 | 9 | 88 | ||||||||
| H-AlPO-5 | 65 | n/a | 60 | 60 | 25 | 22 | 86 | ||||||||
| H-[Al]-Betac | 65 | 14 | 60 | 60 | 99 | 94 | 95 | ||||||||
| ZSM-5-0NC | 70 | n/a | 25 | 50 | 60 | n/a | n/a | ||||||||
| ZSM-5-30NC | 70 | n/a | 25 | 50 | 94 | n/a | n/a | ||||||||
| HUSY | 66 | 2.5 | 10 | 60 | 32 | n/a | n/a | ||||||||
| HUSY | 66 | 15 | 10 | 60 | 90 | n/a | n/a | ||||||||
| HUSY | 66 | 40 | 10 | 60 | 85 | n/a | n/a | ||||||||
| HY | 66 | n/a | 20 | 60 | 10 | n/a | n/a | ||||||||
| Beta | 66 | n/a | 600 | 20 | 89 | n/a | n/a |
References
- Weber, M.M.; Weber, M.M.; Weber, V. Phenol; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2020; pp. 4–7.
- Sheldon, R.A. Homogeneous and Heterogeneous Catalytic Oxidations with Peroxide Reagents. Org. Peroxygen Chem. 1993, 164, 21–43.
- Alekar, N.A.; Indira, V.; Halligudi, S.B.; Srinivas, D.; Gopinathan, S.; Gopinathan, C. Kinetics and Mechanism of Selective Hydroxylation of Benzene Catalysed by Vanadium Substituted Heteropolymolybdates. J. Mol. Catal. A Chem. 2000, 164, 181–189.
- Lee, C.W.; Lee, W.J.; Park, Y.K.; Park, S.E. Catalytic Hydroxylation of Benzene over Vanadium-Containing Molecular Sieves. Catal. Today 2000, 61, 137–141.
- Olah, G.A.; Ohnishi, R. Oxyfunctionalization of Hydrocarbons. 8. Electrophilic Hydroxylation of Benzene, Alkylbenzenes, and Halobenzenes with Hydrogen Peroxide in Superacids. J. Org. Chem. 1978, 43, 865–867.
- Kuznetsova, N.I.; Kuznetsova, L.I.; Kirillova, N.V.; Pokrovskii, L.M.; Detusheva, L.G. Oxidation of Cyclohexene and α Pinene with O2—H2 Mixture. Russ. Chem. Bull. 2003, 52, 1544–1551.
- Niwa, S.I.; Eswaramoorthy, M.; Nair, J.; Raj, A.; Itoh, N.; Shoji, H.; Namba, T.; Mizukami, F. A One-Step Conversion of Benzene to Phenol with a Palladium Membrane. Science 2002, 295, 105–107.
- Sobolev, V.I.; Dubkov, K.A.; Paukshtis, E.A.; Piratko, L.V.; Rodkin, M.A.; Kharitonov, A.S.; Panov, G.I. On the Role of Brønsted Acidity in the Oxidation of Benzene to Phenol by Nitrous Oxide. Appl. Catal. A Gen. 1996, 141, 185–192.
- Dubkov, K.A.; Sobolev, V.I.; Talsi, E.P.; Rodkin, M.A.; Watkins, N.H.; Shteinman, A.A.; Panov, G.I. Kinetic Isotope Effects and Mechanism of Biomimetic Oxidation of Methane and Benzene on FeZSM-5 Zeolite. J. Mol. Catal. A Chem. 1997, 123, 155–161.
- Zakoshansky, V. Phenol Process Celebrates Its 60th Anniversary: The Role of Chemical Principles in Technological Breakthroughs. Russ. J. Gen. Chem. 2009, 79, 2267–2271.
- Morachevskii, A.G.; Nemtsov, M.S. Vospominaniya i Razmyshleniya (Zapiski Khimika) (Recollections and Reflection (Chemist’s Memoirs)). Russ. J. Appl. Chem. 2007, 80, 511–512.
- Hock, H.; Lang, S. Autoxydation von Koblenwasserstoffen: Über Peroxyde von Benzol-Derivaten. Eur. J. Inorg. Chem. 1944, 77, 257–263.
- Udris, R.J.; Sergeyev, P.G.; Kruzhalov, B.D. Sposob Polucheniya Gidroperekisejj Alkilirovannykh-Proizvodnykh Benzola Ili Alicikloaromaticheskikh Uglevodorodov. USSR Patent 106,666, 7 January 1947.
- Yaremenko, I.A.; Vil’, V.A.; Demchuk, D.V.; Terent’ev, A.O. Rearrangements of Organic Peroxides and Related Processes. Beilstein J. Org. Chem. 2016, 12, 1647–1748.
- Duh, Y.S.; Kao, C.S.; Hwang, H.H.; Lee, W.W.L. Thermal Decomposition Kinetics of Cumene Hydroperoxide. Process Saf. Environ. Prot. 1998, 76, 271–276.
- Tsai, H.F.; Guo, S.J.; Wu, S.H. Fire and Thermal Hazard Analyses of Industrial Zeolite Catalysis for Phenol Production. Adv. Mater. Res. 2012, 560–561, 161–166.
- The 100 Largest Losses 1972–2011; Marsh & McLennan: New York, NY, USA, 2011; pp. 1–42.
- Yadav, G.D.; Asthana, N.S. Selective Decomposition of Cumene Hydroperoxide into Phenol and Acetone by a Novel Cesium Substituted Heteropolyacid on Clay. Appl. Catal. A Gen. 2003, 244, 341–357.
- Schmidt, R.J. Industrial Catalytic Processes—Phenol Production. Appl. Catal. A Gen. 2005, 280, 89–103.
- Sheldon, R.A.; van Doorn, J.A. Observation by PMR Spectroscopy of the Intermediate Alkoxycarbonium Ions in the Acid-Catalysed Decomposition of Organic Hydroperoxides. Tetrahedron Lett. 1973, 14, 1021–1022.
- Barton, D.H.R.; Delanghe, N.C. New Catalysts for the Conversion of Cumene Hydroperoxide into Phenol. Tetrahedron Lett. 1997, 38, 73–78.
- Kharasch, M.S.; Fono, A.; Nudenberg, W. The Chemistry Of Hydroperoxides I. The Acid-Catalyzed Decomposition of α,α-Dimethylbenzyl (α-Cumyl) Hydroperxoide. J. Org. Chem. 1950, 15, 748–752.
- Seubold, F.H.; Vaughan, W.E. Acid-Catalyzed Decomposition of Cumene Hydroperoxide. J. Am. Chem. Soc. 1953, 75, 3790–3792.
- Turner, J.O. The Acid-Catalyzed Decomposition of Aliphatic Hydroperoxides: Reactions in the Presence of Alcohols. Tetrahedron Lett. 1971, 12, 887–890.
- Deno, N.C.; Billups, W.E.; Kramer, K.E.; Lastomirsky, R.R. The Rearrangement of Aliphatic Primary, Secondary, and Tertiary Alkyl Hydroperoxides in Strong Acid. J. Org. Chem. 1970, 35, 3080–3082.
- Hock, H.; Kropf, H. Autoxydation von Kohlenwasserstoffen Und Die Cumol-Phenol-Synthese. Angew. Chem. 1957, 69, 313–321.
- Pinnavaia, T.J. Intercalated Clay Catalysts. Science 1983, 220, 365–371.
- Varma, R.S. Clay and Clay-Supported Reagents in Organic Synthesis. Tetrahedron 2002, 58, 1235–1255.
- Theocharis, C.R.; S’Jacob, K.J.; Gray, A.C. Enhancement of Lewis Acidity in Layer Aluminosilicates. Treatment with Acetic Acid. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 1509–1515.
- Cativiela, C.; Fraile, J.M.; Garcia, J.I.; Mayoral, J.A.; Figueras, F.; De Menorval, L.C.; Alonso, P.J. Factors Influencing the K10 Montmorillonite-Catalyzed Diels-Alder Reaction between Methyl Acrylate and Cyclopentadiene. J. Catal. 1992, 137, 394–407.
- Rhodes, C.N.; Brown, D.R. Surface Properties and Porosities of Silica and Acid-Treated Montmorillonite Catalyst Supports: Influence on Activities of Supported ZnCl2 Alkylation Catalysts. J. Chem. Soc. Faraday Trans. 1993, 89, 1387–1391.
- Selvin, R.; Hsu, H.L.; Aneesh, P.; Chen, S.H.; Hung, L.H. Preparation of Acid-Modified Bentonite for Selective Decomposition of Cumene Hydroperoxide into Phenol and Acetone. React. Kinet. Mech. Catal. 2010, 100, 197–204.
- Sasidharan, M.; Kumar, R. Zeolite-Catalysed Selective Decomposition of Cumene Hydroperoxide into Phenol and Acetone. J. Chem. Res.-Part S 1997, 2, 52–53.
- Knifton, J.F.; Sanderson, J.R. Phenol/Acetone Cogeneration via Solid Acid Catalysis. Appl. Catal. A Gen. 1997, 161, 199–211.
- Han, L.; Wang, Y.; Zhang, J.; Lei, Z.; Huang, C.; Chen, B. Acidic Montmorillonite/Cordierite Monolithic Catalysts for Cleavage of Cumene Hydroperoxide. Chin. J. Chem. Eng. 2014, 22, 854–860.
- Pope, M.T. Heteropoly and Isopoly Oxometalates; Springer: Berlin/Heidelberg, Germany, 1993.
- Izumi, Y.; Urabe, K.; Onaka, M. Zeolites, Calys and Heteropoly Acids; VSH Publishers Inc.: London, UK, 1992.
- Okuhara, T.; Mizuno, N.; Misono, M. Catalytic Chemistry of Heteropoly Compounds. Adv. Catal. 1996, 41, 113–252.
- Misono, M.; Nojiri, N. Recent Progress in Catalytic Technology in Japan. Appl. Catal. 1990, 64, 1–30.
- Corma, A. Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions. Chem. Rev. 1995, 95, 559–614.
- Kozhevnikov, I.V. Heteropoly Acids as Catalysts for Fine Chemicals Synthesis; Woodhead Publishing Ltd.: Sawston, UK, 2005; Volume 6.
- Chang, C.D.; Pelrine, P.P. Mobil Oil Corporation Production of Phenol. U.S. Patent 4490565A, 25 December 1984.
- Romano, U.; Clerici, M.G.; Bellussi, G.; Buonomo, F. Catalyst for the Selective Decomposition of Cumene Hydroperoxide. U.S. Patent 47543573A, 10 May 1988.
- Romano, U.; Clerici, M.G.; Bellussi, G.; Buonomo, F. Catalyst for the Selective Decompostion of Cumene Hydroperoxide and Process Using it. U.S. Patent 4849387A, 18 July 1989.
- Xu, R.; Pang, W.; Yu, J.; Huo, Q.; Chen, J. Chemistry of Zeolites and Related Porous Materials; Wiley: Hoboken, NJ, USA, 2007.
- Cejka, J.; Corma, A.; Zones, S. Zeolites and Catalysis; Wiley: Hoboken, NJ, USA, 2010.
- Koltunov, K.Y.; Sobolev, V.I. Efficient Cleavage of Cumene Hydroperoxide over HUSY Zeolites: The Role of Brønsted Acidity. Appl. Catal. A Gen. 2008, 336, 29–34.
- Kumar, K.P.; Selvin, R.; Kumari, P.; Roselin, L.S.; Arul, N.S.; Bououdina, M. Selective Decomposition of Cumene Hydroperoxide into Phenol and Acetone over Nanocrystalline ZSM-5. Int. J. Mater. Eng. Innov. 2010, 1, 417–425.
- Hedge, S.G.; Ratnasamy, P.; Kustov, L.M.; Kazansky, V.B. Acidity and Catalytic Activity of SAPO-5 and AlPO-5 Molecular Sieves. Zeolites 1988, 8, 137–141.
- Chaudhary, V.R.; Singh, A.P.; Kumar, R. Acidity and Sorbate Shape Selectivity of H-ZSM-22, H-ZSM-48, and H-ZSM-50 Zeolites. J. Catal. 1991, 129, 293–296.
- Guisnet, M.; Gilson, J.-P. Zeolites for Cleaner Technologies; Imperial College Press: London, UK, 2002.
- Louis, B.; Walspurger, S.; Sommer, J. Quantitative Determination of Brönsted Acid Sites on Zeolites: A New Approach towards the Chemical Composition of Zeolites. Catal. Lett. 2004, 93, 81–84.
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Superelectrophilic Activation of Polyfunctional Organic Compounds Using Zeolites and Other Solid Acids. Chem. Commun. 2004, 15, 1754.
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Cyclization of 1-Phenyl-2-Propen-1-Ones into 1-Indanones Using H-Zeolite and Other Solid Acids. The Role of Mono- and Dicationic Intermediates. Tetrahedron Lett. 2005, 46, 8391–8393.
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Selective, C,C-Double Bond Reduction of α,β-Unsaturated Carbonyl Compounds with Cyclohexane Using Zeolites. J. Mol. Catal. A Chem. 2006, 245, 231–234.
- Ye, J.; Li, J.; Sha, Y.; Xu, Y.; Zhou, D. Novel Reactive Distillation Process for Phenol Production with a Dry Cation Exchange Resin as the Catalyst. Ind. Eng. Chem. Res. 2014, 53, 12614–12621.
- Huang, D.; Han, M.; Wang, J.; Jin, Y. Catalytic Decomposition Process of Cumene Hydroperoxide Using Sulfonic Resins as Catalyst. Chem. Eng. J. 2002, 88, 215–223.