Aging is characterized by the dynamic remodeling of the immune system designated “immunosenescence,” and is associated with altered hematopoiesis, thymic involution, and lifelong immune stimulation by multitudinous chronic stressors, including the cytomegalovirus (CMV). Such alterations may contribute to a lowered proportion of naïve T-cells and to reduced diversity of the T-cell repertoire. In the peripheral circulation, a shift occurs towards accumulations of T and B-cell populations with memory phenotypes, and to accumulation of putatively senescent and exhausted immune cells. The aging-related accumulations of functionally exhausted memory T lymphocytes, commonly secreting pro-inflammatory cytokines, together with mediators and factors of the innate immune system, are considered to contribute to the low-grade inflammation (inflammaging) often observed in elderly people. These senescent immune cells not only secrete inflammatory mediators, but are also able to negatively modulate their environments. In this review, we give a short summary of the ways that immunosenescence, inflammaging, and CMV infection may cause insufficient immune responses, contribute to the establishment of the hyperinflammatory syndrome and impact the severity of the coronavirus disease 2019 (COVID-19) in elderly people.
- COVID-19
- immunosenescence
- inflammaging
- cytokine storm
- CMV
- hyperinflammatory syndrome
1. Introduction
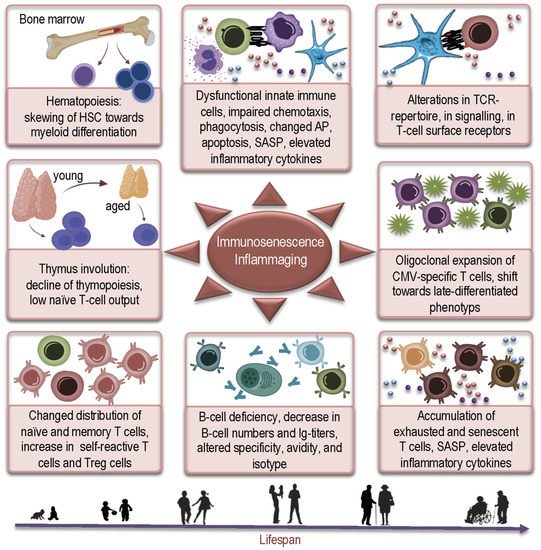
2. How Aged Bone Marrow and Thymic Aging May Contribute to Susceptibility to COVID-19
2.1. Age-Related Changes in Hematopoiesis and COVID-19
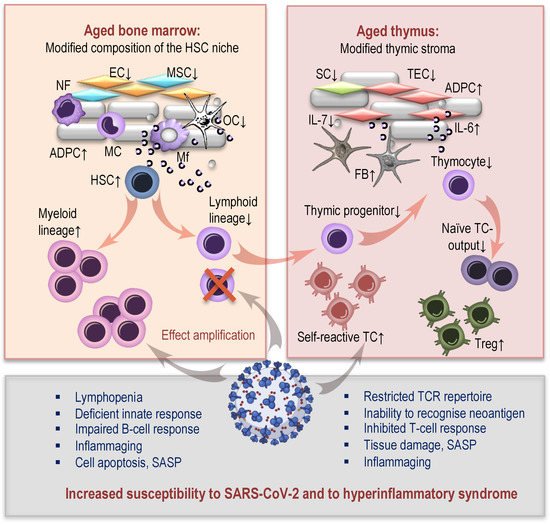
2.2. Thymic Aging and COVID-19
3. How Age-Related Changes in Innate and Adaptive Immunity Induce Inflammaging and May Influence Susceptibility to SARS-CoV-2 and Severity of COVID-19
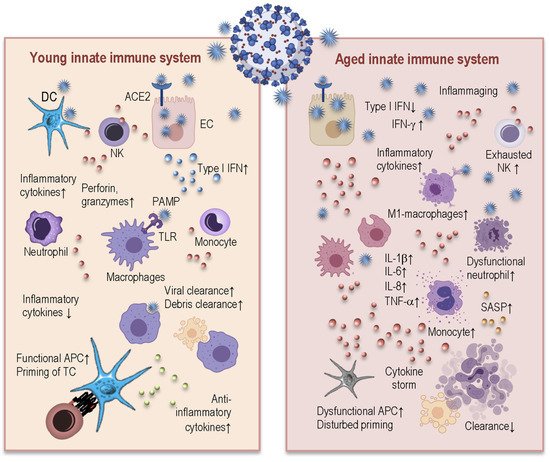
3.1. Age-Related Changes in Innate Immunity and Their Consequences for COVID-19
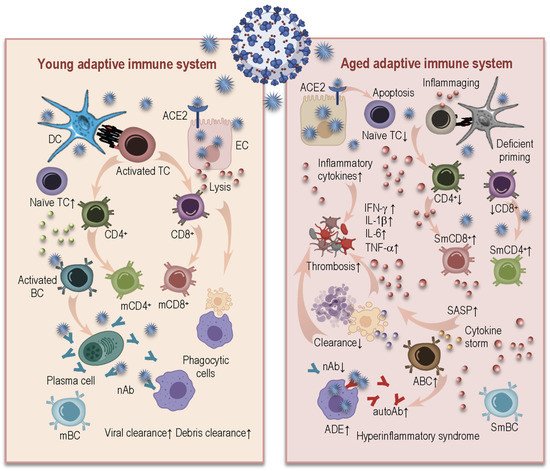
3.2. Age-Related Alterations in Adaptive Immunity and COVID-19
References
- Weng, N.P. Aging of the immune system: How much can the adaptive immune system adapt? Immunity 2006, 24, 495–499.
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247.
- Pawelec, G. Age and immunity: What is “immunosenescence”? Exp. Gerontol. 2018, 105, 4–9.
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. NY Acad. Sci. 2000, 908, 244–254.
- Wang, J.; Geiger, H.; Rudolph, K.L. Immunoaging induced by hematopoietic stem cell aging. Curr. Opin. Immunol. 2011, 23, 532–536.
- Aspinall, R.; Andrew, D. Thymic involution in aging. J. Clin. Immunol. 2000, 20, 250–256.
- Weiskopf, D.; Weinberger, B.; Grubeck-Loebenstein, B. The aging of the immune system. Transpl. Int. 2009, 22, 1041–1050.
- Shaw, A.C.; Joshi, S.; Greenwood, H.; Panda, A.; Lord, J.M. Aging of the innate immune system. Curr. Opin. Immunol. 2010, 22, 507–513.
- Fulop, T.; Larbi, A.; Douziech, N.; Fortin, C.; Guerard, K.P.; Lesur, O.; Khalil, A.; Dupuis, G. Signal transduction and functional changes in neutrophils with aging. Aging Cell 2004, 3, 217–226.
- Fernandez-Morera, J.L.; Calvanese, V.; Rodriguez-Rodero, S.; Menendez-Torre, E.; Fraga, M.F. Epigenetic regulation of the immune system in health and disease. Tissue Antigens 2010, 76, 431–439.
- Gonzalo, S. Epigenetic alterations in aging. J. Appl. Physiol. 2010, 109, 586–597.
- Müller, L.; Pawelec, G. Aging and immunity-impact of behavioral intervention. Brain Behav. Immun. 2014, 39, 8–22.
- Alves, A.S.; Bueno, V. Immunosenescence: Participation of T lymphocytes and myeloid-derived suppressor cells in aging-related immune response changes. Einstein (Sao Paulo) 2019, 17, eRB4733.
- Zhang, H.; Weyand, C.M.; Goronzy, J.J. Hallmarks of the aging T-cell system. FEBS J. 2021.
- Müller, L.; Hamprecht, K.; Pawelec, G. The role of CMV in “immunosenescence”. In The Ageing Immune System and Health; Bueno, V., Lord, J.M., Jackson, T.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 53–68.
- Weltevrede, M.; Eilers, R.; de Melker, H.E.; van Baarle, D. Cytomegalovirus persistence and T-cell immunosenescence in people aged fifty and older: A systematic review. Exp. Gerontol. 2016, 77, 87–95.
- Karrer, U.; Sierro, S.; Wagner, M.; Oxenius, A.; Hengel, H.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Memory inflation: Continuous accumulation of antiviral CD8+ T cells over time. J. Immunol. 2003, 170, 2022–2029.
- Kim, J.; Kim, A.R.; Shin, E.C. Cytomegalovirus Infection and Memory T Cell Inflation. Immune Netw. 2015, 15, 186–190.
- Ouyang, Q.; Wagner, W.M.; Voehringer, D.; Wikby, A.; Klatt, T.; Walter, S.; Muller, C.A.; Pircher, H.; Pawelec, G. Age-associated accumulation of CMV-specific CD8+ T cells expressing the inhibitory killer cell lectin-like receptor G1 (KLRG1). Exp. Gerontol. 2003, 38, 911–920.
- Pawelec, G.; Derhovanessian, E.; Larbi, A.; Strindhall, J.; Wikby, A. Cytomegalovirus and human immunosenescence. Rev. Med Virol. 2009, 19, 47–56.
- Pawelec, G.; McElhaney, J.E.; Aiello, A.E.; Derhovanessian, E. The impact of CMV infection on survival in older humans. Curr. Opin. Immunol. 2012, 24, 507–511.
- Pawelec, G.; Derhovanessian, E. Role of CMV in immune senescence. Virus Res. 2011, 157, 175–179.
- Savva, G.M.; Pachnio, A.; Kaul, B.; Morgan, K.; Huppert, F.A.; Brayne, C.; Moss, P.A.; Medical Research Council Cognitive, F.; Ageing, S. Cytomegalovirus infection is associated with increased mortality in the older population. Aging Cell 2013, 12, 381–387.
- Wikby, A.; Ferguson, F.; Forsey, R.; Thompson, J.; Strindhall, J.; Lofgren, S.; Nilsson, B.O.; Ernerudh, J.; Pawelec, G.; Johansson, B. An immune risk phenotype, cognitive impairment, and survival in very late life: Impact of allostatic load in Swedish octogenarian and nonagenarian humans. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 556–565.
- Spyridopoulos, I.; Martin-Ruiz, C.; Hilkens, C.; Yadegarfar, M.E.; Isaacs, J.; Jagger, C.; Kirkwood, T.; von Zglinicki, T. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: Results from the Newcastle 85+ study. Aging Cell 2015.
- Colonna-Romano, G.; Bulati, M.; Aquino, A.; Vitello, S.; Lio, D.; Candore, G.; Caruso, C. B cell immunosenescence in the elderly and in centenarians. Rejuvenation Res. 2008, 11, 433–439.
- Ademokun, A.; Wu, Y.C.; Dunn-Walters, D. The ageing B cell population: Composition and function. Biogerontology 2010, 11, 125–137.
- Dewan, S.K.; Zheng, S.B.; Xia, S.J.; Bill, K. Senescent remodeling of the immune system and its contribution to the predisposition of the elderly to infections. Chin. Med. J. 2012, 125, 3325–3331.
- Costagliola, G.; Spada, E.; Consolini, R. Age-related differences in the immune response could contribute to determine the spectrum of severity of COVID-19. Immun. Inflamm. Dis. 2021, 9, 331–339.
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116.
- Pietras, E.M.; Reynaud, D.; Kang, Y.A.; Carlin, D.; Calero-Nieto, F.J.; Leavitt, A.D.; Stuart, J.M.; Gottgens, B.; Passegue, E. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 2015, 17, 35–46.
- Compston, J.E. Bone marrow and bone: A functional unit. J. Endocrinol. 2002, 173, 387–394.
- Gruver, A.L.; Hudson, L.L.; Sempowski, G.D. Immunosenescence of ageing. J. Pathol. 2007, 211, 144–156.
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590.
- Dykstra, B.; de Haan, G. Hematopoietic stem cell aging and self-renewal. Cell Tissue Res 2008, 331, 91–101.
- Warren, L.A.; Rossi, D.J. Stem cells and aging in the hematopoietic system. Mech. Ageing Dev. 2009, 130, 46–53.
- Konieczny, J.; Arranz, L. Updates on Old and Weary Haematopoiesis. Int. J. Mol. Sci. 2018, 19, 2567.
- Geiger, H.; de Haan, G.; Florian, M.C. The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol. 2013, 13, 376–389.
- Jasiulionis, M.G. Abnormal Epigenetic Regulation of Immune System during Aging. Front. Immunol. 2018, 9, 197.
- Müller, L.; Di Benedetto, S.; Pawelec, G. The Immune System and Its Dysregulation with Aging. Subcell. Biochem. 2019, 91, 21–43.
- Müller, L.; Pawelec, G. As we age: Does slippage of quality control in the immune system lead to collateral damage? Ageing Res. Rev. 2015, 23, 116–123.
- Aspinall, R.; Pitts, D.; Lapenna, A.; Mitchell, W. Immunity in the elderly: The role of the thymus. J. Comp. Pathol. 2010, 142 (Suppl. S1), S111–S115.
- Kugelberg, E. Immunometabolism: Unravelling the puzzle to longevity and immunity. Nat. Rev. Immunol. 2016, 16, 74–75.
- Sempowski, G.D.; Hale, L.P.; Sundy, J.S.; Massey, J.M.; Koup, R.A.; Douek, D.C.; Patel, D.D.; Haynes, B.F. Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J. Immunol. 2000, 164, 2180–2187.
- Mitchell, W.A.; Lang, P.O.; Aspinall, R. Tracing thymic output in older individuals. Clin. Exp. Immunol. 2010, 161, 497–503.
- Wang, W.; Thomas, R.; Oh, J.; Su, D.M. Thymic Aging May Be Associated with COVID-19 Pathophysiology in the Elderly. Cells 2021, 10, 628.
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2.
- Oh, J.; Wang, W.; Thomas, R.; Su, D.M. Capacity of tTreg generation is not impaired in the atrophied thymus. PLoS Biol. 2017, 15, e2003352.
- Raynor, J.; Lages, C.S.; Shehata, H.; Hildeman, D.A.; Chougnet, C.A. Homeostasis and function of regulatory T cells in aging. Curr. Opin. Immunol. 2012, 24, 482–487.
- Chougnet, C.A.; Tripathi, P.; Lages, C.S.; Raynor, J.; Sholl, A.; Fink, P.; Plas, D.R.; Hildeman, D.A. A major role for Bim in regulatory T cell homeostasis. J. Immunol. 2011, 186, 156–163.
- Bartleson, J.M.; Radenkovic, D.; Covarrubias, A.J.; Furman, D.; Winer, D.A.; Verdin, E. SARS-CoV-2, COVID-19 and the aging immune system. Nat. Aging 2021, 1, 769–782.
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114.
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469.
- Zheng, Y.; Liu, X.; Le, W.; Xie, L.; Li, H.; Wen, W.; Wang, S.; Ma, S.; Huang, Z.; Ye, J.; et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell 2020, 11, 740–770.
- Stahl, E.C.; Brown, B.N. Cell Therapy Strategies to Combat Immunosenescence. Organogenesis 2015, 11, 159–172.
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 533–535.
- Masselli, E.; Vaccarezza, M.; Carubbi, C.; Pozzi, G.; Presta, V.; Mirandola, P.; Vitale, M. NK cells: A double edge sword against SARS-CoV-2. Adv. Biol. Regul. 2020, 77, 100737.
- Van Eeden, C.; Khan, L.; Osman, M.S.; Cohen Tervaert, J.W. Natural Killer Cell Dysfunction and Its Role in COVID-19. Int. J. Mol. Sci. 2020, 21, 6351.
- Zhou, R.; To, K.K.; Wong, Y.C.; Liu, L.; Zhou, B.; Li, X.; Huang, H.; Mo, Y.; Luk, T.Y.; Lau, T.T.; et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity 2020, 53, 864–877.
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.G.; Dubuisson, A.; Derosa, L.; Almire, C.; Henon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Bassler, K.; Schlickeiser, S.; Zhang, B.; Kramer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.
- Seidler, S.; Zimmermann, H.W.; Bartneck, M.; Trautwein, C.; Tacke, F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010, 11, 30.
- Wong, C.; Goldstein, D.R. Impact of aging on antigen presentation cell function of dendritic cells. Curr. Opin. Immunol. 2013, 25, 535–541.
- Hazeldine, J.; Lord, J.M. Immunesenescence: A Predisposing Risk Factor for the Development of COVID-19? Front. Immunol. 2020, 11, 573662.
- Vallejo, A.N. CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol. Rev. 2005, 205, 158–169.
- Pangrazzi, L.; Weinberger, B. T cells, aging and senescence. Exp. Gerontol. 2020, 134, 110887.
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448.
- Boucher, N.; Dufeu-Duchesne, T.; Vicaut, E.; Farge, D.; Effros, R.B.; Schachter, F. CD28 expression in T cell aging and human longevity. Exp. Gerontol. 1998, 33, 267–282.
- Czesnikiewicz-Guzik, M.; Lee, W.W.; Cui, D.; Hiruma, Y.; Lamar, D.L.; Yang, Z.Z.; Ouslander, J.G.; Weyand, C.M.; Goronzy, J.J. T cell subset-specific susceptibility to aging. Clin. Immunol. 2008, 127, 107–118.
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118.
- Ciabattini, A.; Garagnani, P.; Santoro, F.; Rappuoli, R.; Franceschi, C.; Medaglini, D. Shelter from the cytokine storm: Pitfalls and prospects in the development of SARS-CoV-2 vaccines for an elderly population. Semin. Immunopathol. 2020, 42, 619–634.
- Van Eijk, L.E.; Binkhorst, M.; Bourgonje, A.R.; Offringa, A.K.; Mulder, D.J.; Bos, E.M.; Kolundzic, N.; Abdulle, A.E.; van der Voort, P.H.; Olde Rikkert, M.G.; et al. COVID-19: Immunopathology, pathophysiological mechanisms, and treatment options. J. Pathol. 2021, 254, 307–331.
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108.
- Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19.
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.
- Juno, J.A.; Tan, H.X.; Lee, W.S.; Reynaldi, A.; Kelly, H.G.; Wragg, K.; Esterbauer, R.; Kent, H.E.; Batten, C.J.; Mordant, F.L.; et al. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat. Med. 2020, 26, 1428–1434.
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. The generation of memory B cells is maintained, but the antibody response is not, in the elderly after repeated influenza immunizations. Vaccine 2016, 34, 2834–2840.
- Zhu, Z.; Chakraborti, S.; He, Y.; Roberts, A.; Sheahan, T.; Xiao, X.; Hensley, L.E.; Prabakaran, P.; Rockx, B.; Sidorov, I.A.; et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2007, 104, 12123–12128.
- Cox, R.J.; Brokstad, K.A. Not just antibodies: B cells and T cells mediate immunity to COVID-19. Nat. Rev. Immunol. 2020, 20, 581–582.
- Kaneko, N.; Kuo, H.H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183, 143–157.
- Bruunsgaard, H.; Andersen-Ranberg, K.; Hjelmborg, J.; Pedersen, B.K.; Jeune, B. Elevated levels of tumor necrosis factor alpha and mortality in centenarians. Am. J. Med. 2003, 115, 278–283.
- Bulati, M.; Buffa, S.; Candore, G.; Caruso, C.; Dunn-Walters, D.K.; Pellicano, M.; Wu, Y.C.; Colonna Romano, G. B cells and immunosenescence: A focus on IgG+IgD-CD27- (DN) B cells in aged humans. Ageing Res. Rev. 2011, 10, 274–284.
- Weksler, M.E. Changes in the B-cell repertoire with age. Vaccine 2000, 18, 1624–1628.
- Ratliff, M.; Alter, S.; Frasca, D.; Blomberg, B.B.; Riley, R.L. In senescence, age-associated B cells secrete TNFalpha and inhibit survival of B-cell precursors. Aging Cell 2013, 12, 303–311.
- Cancro, M.P. Age-Associated B Cells. Annu. Rev. Immunol. 2020, 38, 315–340.
- Frasca, D. Senescent B cells in aging and age-related diseases: Their role in the regulation of antibody responses. Exp. Gerontol. 2018, 107, 55–58.
- Woodruff, M.C.; Ramonell, R.P.; Nguyen, D.C.; Cashman, K.S.; Saini, A.S.; Haddad, N.S.; Ley, A.M.; Kyu, S.; Howell, J.C.; Ozturk, T.; et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 2020, 21, 1506–1516.
- Ricke, D.O. Two Different Antibody-Dependent Enhancement (ADE) Risks for SARS-CoV-2 Antibodies. Front. Immunol. 2021, 12, 640093.
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, Immunity, and COVID-19: How Age Influences the Host Immune Response to Coronavirus Infections? Front. Physiol. 2020, 11, 571416.