Killer yeasts produce protein toxins that are lethal to sensitive yeasts. The synthesis and secretion of killer toxins by Torulaspora delbrueckii (Td) and Saccharomyces cerevisiae (Sc) requires the presence of at least two cytoplasmic dsRNA viruses that are members of the family Totiviridae. One is a satellite virus with a medium-size genome (V-M) that encodes the toxin, and the other is a helper virus with a large-size genome ((V-LA or V-LBC)) that provides the capsid and polymerase required for maintenance and replication of both viruses. The structure, origin and putative functions of different sequences on the genome of TdV-LBC are analyzed here.
- Torulaspora
- killer
- LBC virus
- dsRNA genome
- high-throughput sequencing (HTS)
- frameshifting nucleotide insertions or deletions (indels)
- xenolog
1. Introduction: Yeast Double-Stranded RNA viruses
The yeasts Torulaspora delbrueckii (Td) and Saccharomyces cerevisiae (Sc) may show a killer phenotype that is encoded in dsRNA M viruses (V-M), which require the helper activity of another dsRNA virus (V-LA or V-LBC) for replication. Recently, two TdV-LBCbarr genomes, which share sequence identity with ScV-LBC counterparts, were characterized by high-throughput sequencing (HTS). They also share some similar characteristics with Sc-LA viruses. This may explain why TdV-LBCbarr has helper capability to maintain M viruses, whereas ScV-LBC does not.A specific LA virus may support different types of satellite M viruses but usually only one type in each killer yeast strain [1][2]. LBC viruses are another type of large-size dsRNA virus that may coexist with V-LA and V-M in the cytoplasm of S. cerevisiae; although no helper activity is known for V-LBC in this yeast species [3][4][5][6][7]. However, a new LBC virus has recently been found in T. delbrueckii (TdV-LBCbarr2) that may act as a helper for two M viruses in the same yeast strain: TdV-Mbarr1 and ScV-M1 [8]. These viruses are inherited in the cytoplasm from mother yeast to daughter bud and transferred horizontally between different yeasts by mating or heterokaryon formation [9]. However, the Sc-M1 virus has been recently found in Td, which suggests that it may have been transferred horizontally between different yeasts by conventional viral infection [8].
2. Canonical Nucleotide and Amino Acid Sequences of Td-LBC and Sc-LBC Viruses
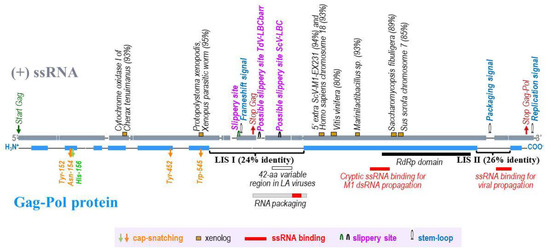
Figure 1. A schematic representation of TdV-LBCbarr (ssRNA and Gag-Pol) showing the two stretches of low sequence identity with Gag-Pol of ScV-LBC1-original: stretch I (LIS I) and stretch II (LIS II). Grey (ssRNA) and blue (Gag-Pol protein) shaded regions are areas of significant identity (above 50% in windows of 50 nucleotides or 20 amino acids in length) between TdV-LBCbarr2 and ScV-LBC1-original genomes. Global alignment was done using Clone Manager 7.11. Scoring matrix: Linear (Mismatch 2, OpenGap 4, ExtGap 1 for cDNA; and BLOSUM 62 for protein). Relevant RNA codons and motifs of the viral genome are displayed above the (+)ssRNA, as well as stretches (≥30 nucleotides) showing high local identity (≥80%) with xenolog sequences of cellular organisms (percentage of identity is in parenthesis). Relevant amino acids for cap-snatching and RdRp domain are shown below Gag-Pol, as well as the location of 42-amino acid variable region, ssRNA binding regions, and RNA packaging region previously described in yeast LA viruses.
3. Features Found in the 5′- and 3′-Extra Sequences from LBC Genomes
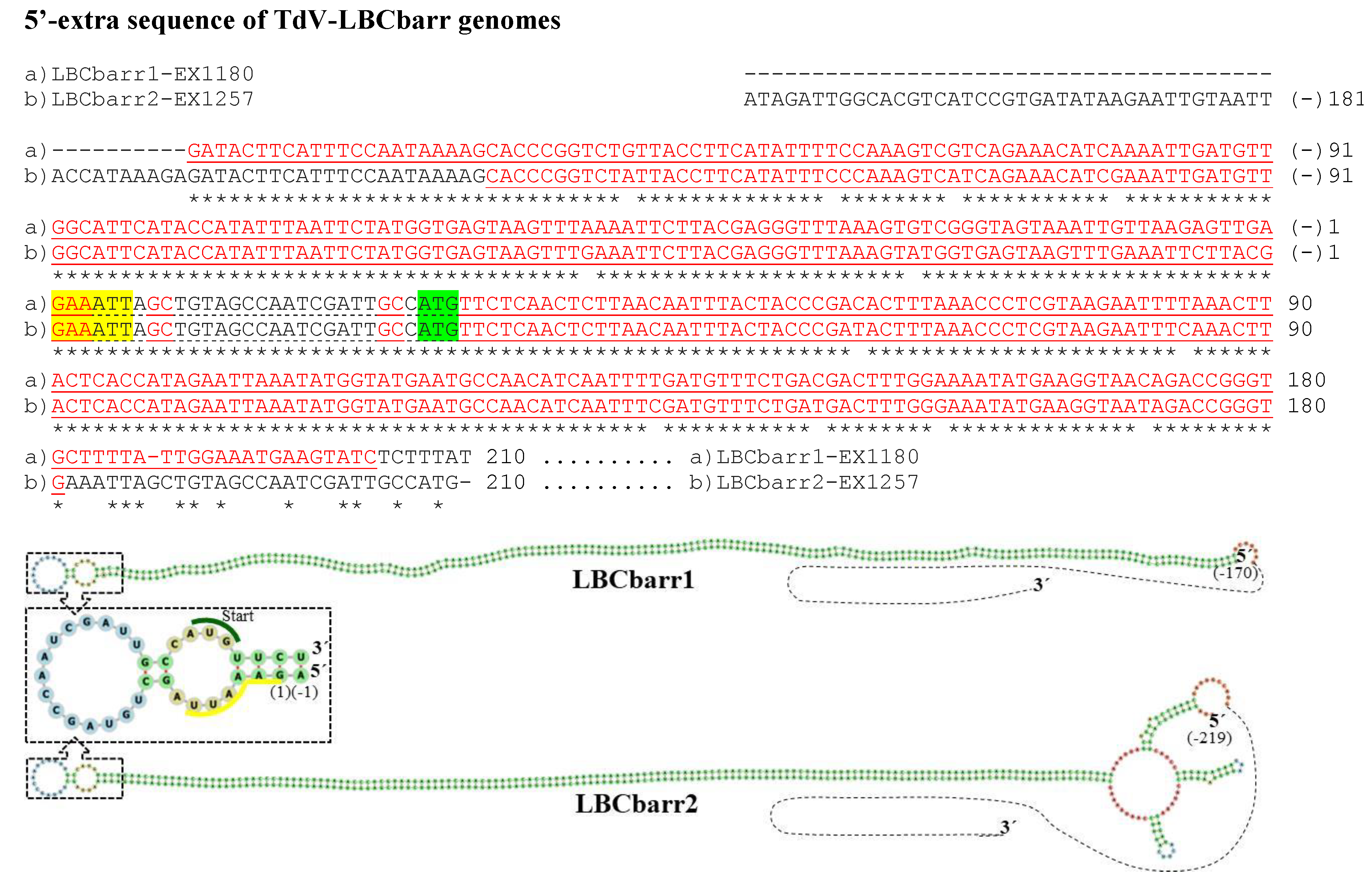
Figure 2. cDNA of 5′-extra sequences and proximal canonical sequences of TdV-LBCbarr1 and TdV-LBCbarr2 genomes. The 5′-GAAATT end of the canonical sequence is yellow highlighted. The protein synthesis initiation codon of Gag-Pol is green shaded. Nucleotides of palindromic sequences are shown in red letters. The stem-loop sequences are underlined. The unpaired nucleotides of each loop are dot underlined. Asterisks (*) indicate identical nucleotides. The secondary RNA structures of possible 5′ stem-loops are shown at the bottom of the sequences.
No relevant identity was detected between the 5′- or 3′-extra sequences of V-LBC genomes from T. delbrueckiiand S. cerevisiae. However, high local identity between the 5′- or 3′-extra sequences from Td-LBCbarr viruses, and between the 5′-extra sequences from Sc-LBC viruses was found (Figure 2, Figure 3 and Figure 4). No relevant identity between the 3′-extra sequences from Sc-LBC viruses was found.
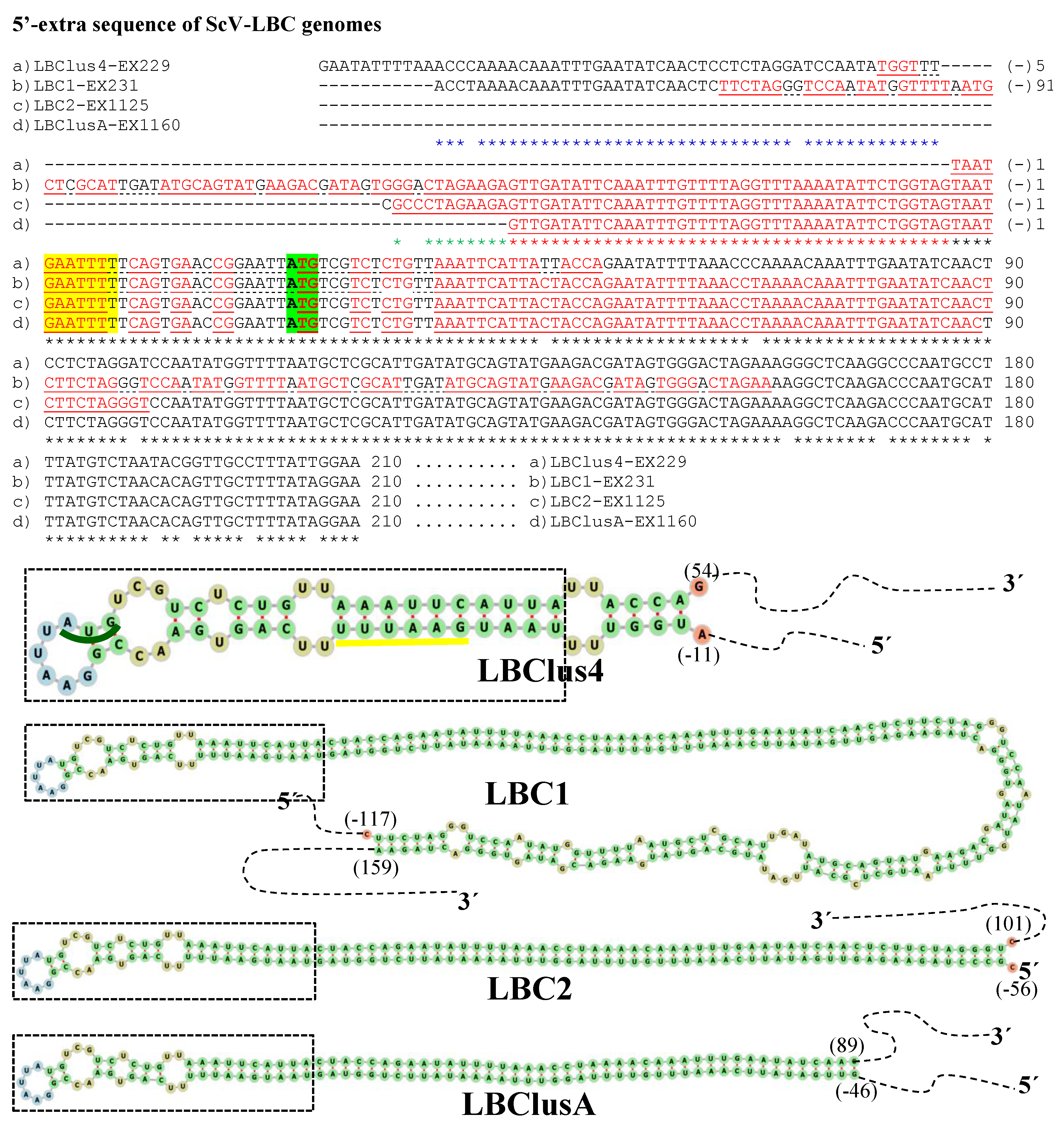
Figure 3. Nucleotide sequence (cDNA) alignment of 5′-extra sequences and proximal canonical sequences of ScV-LBC genomes from S. cerevisiae: (a) LBClus4-EX229, (b) LBC1-EX231, (c) LBC2-EX1125, and (d) LBClusA-EX1160. Black asterisks (*) indicate identical nucleotides in all genomes. Red asterisks (*) indicate identical nucleotides in (b) LBC1-EX231, (c) LBC2-EX1125, and (d) LBClusA-EX1160 genomes. Blue asterisks (*) indicate identical nucleotides in (a) LBClus4-EX229 and (b) LBC1-EX231 genomes. Green asterisks (*) indicate identical nucleotides in (b) LBC1-EX231 and (c) LBC2-EX1125 genomes. The 5′-GAATTTT of canonical sequences is yellow highlighted. The protein synthesis initiation codon of Gag-Pol is green shaded. Nucleotides of palindromic sequences are shown in red letters. Stem-loops are underlined. The unpaired nucleotides of each loop are dot underlined. The secondary RNA structures of possible 5′ stem-loops are shown at the bottom of the sequences.

Figure 4. cDNA of 3′-extra sequences and proximal canonical sequences of TdV-LBCbarr1-EX1180 and TdV-LBCbarr2-EX1257 genomes. The CAACGGC-3′ ends of canonical sequences are highlighted. The protein synthesis stop codons of Gag-Pol are red-shaded. Nucleotides of palindromic sequences are shown in red. Stem-loops are underlined. The unpaired nucleotides of each loop are dot underlined. Asterisks (*) indicate identical nucleotides. The secondary RNA structures of possible 3′ stem-loops are shown at the bottom of the sequences.
4. Conclusions
The two LIS found in TdV-LBCbarr Gag-Pol may have been originated by successive indels that allow virus speciation while maintaining the fundamental functions of (+) RNA, and Gag and Pol domains. The existence of a second in-frame translation re-initiation codon, preceded by a possible slippery site, may facilitate a required “compensatory frameshift”. This strategy would allow LBC viruses to change their ability to bind newly arisen versions of M (+)RNA for packaging and replication. The transfer of xenolog RNA sequence stretches from different organisms to the canonical sequence of V-L genomes could be at the inception of these viruses. The extra sequences located at both sides of V-LBC canonical genomes may form RNA secondary structures involved in avoiding ssRNA degradation and facilitating dsRNA synthesis, or in a still unknown biological function related to virus replication. The finding of rRNA stretches in the 3′-extra sequences of ScV-LBC genomes may be a consequence of recombination of virus RNA with yeast rRNA. This could form a kind of ribonucleoprotein that somehow resembles the yeast ribosome and ensure the permanence of these viruses in the yeast cell.References
- Schmitt, M.J.; Breinig, F. Yeast viral killer toxins: Lethality and self-protection. Nat. Rev. Microbiol. 2006, 4, 212–221.
- Wickner, R.B.; Fujimura, T.; Esteban, R. Viruses and Prions of Saccharomyces cerevisiae. Adv. Appl. Microbiol. 2013, 86, 1–36.
- Ramírez, M.; Velázquez, R.; López-Piñeiro, A.; Naranjo, B.; Roig, F.; Llorens, C. New Insights into the Genome Organization of Yeast Killer Viruses Based on “Atypical” Killer Strains Characterized by High-Throughput Sequencing. Toxins 2017, 9, 292.
- Rodríguez-Cousiño, N.; Esteban, R. Relationships and Evolution of Double-Stranded RNA Totiviruses of Yeasts Inferred from Analysis of L-A-2 and L-BC Variants in Wine Yeast Strain Populations. Appl. Environ. Microbiol. 2017, 83, e02991-16.
- Rodríguez-Cousiño, N.; Maqueda, M.; Ambrona, J.; Zamora, E.; Esteban, E.; Ramírez, M. A new wine Saccharomyces cerevisiae double-stranded RNA virus encoded killer toxin (Klus) with broad antifungal activity is evolutionarily related to a chromosomal host gene. Appl. Environ. Microbiol. 2011, 77, 1822–1832.
- Vepštaitė-Monstavičė, I.; Lukša, J.; Konovalovas, A.; Ežerskytė, D.; Stanevičienė, R.; Strazdaitė-Žielienė, Ž.; Serva, S.; Servienė, E. Saccharomyces paradoxus K66 Killer System Evidences Expanded Assortment of Helper and Satellite Viruses. Viruses 2018, 10, 564.
- Park, C.-M.; Lopinski, J.D.; Masuda, J.; Tzeng, T.-H.; Bruenn, J. A Second Double-Stranded RNA Virus from Yeast. Virology 1996, 216, 451–454.
- Ramírez, M.; Velázquez, R.; López-Piñeiro, A.; Martínez, A. Genome Features of a New Double-Stranded RNA Helper Virus (LBCbarr) from Wine Torulaspora delbrueckii Killer Strains. Int. J. Mol. Sci. 2021, 22, 13492.
- Wickner, R.B. Double-stranded RNA viruses of Saccharomyces cerevisiae. Microbiol. Rev. 1996, 60, 250–265.
- Dinman, J.D.; Wickner, R.B. Ribosomal frameshifting efficiency and gag/gag-pol ratio are critical for yeast M1 double-stranded RNA virus propagation. J. Virol. 1992, 66, 3669–3676.
- Fujimura, T.; Ribas, J.; Makhov, A.M.; Wickner, R.B. Pol of gag–pol fusion protein required for encapsidation of viral RNA of yeast L-A virus. Nature 1992, 359, 746–749.
- Icho, T.; Wickner, R.B. The Double-stranded RNA Genome of Yeast Virus L-A Encodes Its Own Putative RNA Polymerase by Fusing Two Open Reading Frames. J. Biol. Chem. 1989, 264, 6716–6723.
- Ramírez, M.; Velázquez, R.; Maqueda, M.; Martínez, A. Genome Organization of a New Double-Stranded RNA LA Helper Virus from Wine Torulaspora delbrueckii Killer Yeast as Compared with Its Saccharomyces Counterparts. Front. Microbiol. 2020, 11, 2977.
- Rodríguez-Cousiño, N.; Gómez, P.; Esteban, R. L-A-lus, a New Variant of the L-A Totivirus Found in Wine Yeasts with Klus Killer Toxin-Encoding Mlus Double-Stranded RNA: Possible Role of Killer Toxin-Encoding Satellite RNAs in the Evolution of Their Helper Viruses. Appl. Environ. Microbiol. 2013, 79, 4661–4674.
- Fujimura, T.; Esteban, R.; Esteban, L.M.; Wickner, R.B. Portable encapsidation signal of the L-A double-stranded-RNA virus of Saccharomyces cerevisiae. Cell 1990, 62, 819–828.
- Ramírez, M.; Velázquez, R.; Maqueda, M.; López-Piñeiro, A.; Ribas, J.C. A new wine Torulaspora delbrueckii killer strain with broad antifungal activity and its toxin-encoding double-stranded RNA virus. Front. Microbiol. 2015, 6, 983.
- Rodríguez-Cousiño, N.; Gómez, P.; Esteban, R. Variation and Distribution of L-A Helper Totiviruses in Saccharomyces sensu stricto Yeasts Producing Different Killer Toxins. Toxins 2017, 9, 313.
- Thiele, D.J.; Leibowitz, M.J. Structural and functional analysis of separated strands of killer double-stranded RNA of yeast. Nucleic Acids Res. 1982, 10, 6903–6918.
- Thiele, D.J.; Wang, R.W.; Leibowitz, M.J. Separation and sequence of the 3′ termini of M double-stranded RNA from killer yeast. Nucleic Acids Res. 1982, 10, 1661–1678.
- Park, D.; Hahn, Y. Rapid protein sequence evolution via compensatory frameshift is widespread in RNA virus genomes. BMC Bioinform. 2021, 22, 251.
- Lim, C.S.; Brown, C.M. Know Your Enemy: Successful Bioinformatic Approaches to Predict Functional RNA Structures in Viral RNAs. Front. Microbiol. 2018, 8, 2582.
- Gmyl, A.P.; Korshenko, S.A.; Belousov, E.V.; Khitrina, E.V.; Agol, V.I. Nonreplicative homologous RNA recombination: Promiscuous joining of RNA pieces? RNA 2003, 9, 1221–1231.
- Sztuba-Solińska, J.; Urbanowicz, A.; Figlerowicz, M.; Bujarski, J.J. RNA-RNA Recombination in Plant Virus Replication and Evolution. Annu. Rev. Phytopathol. 2011, 49, 415–443.
- Flynn, P.J.; Moreau, C.S. Assessing the Diversity of Endogenous Viruses Throughout Ant Genomes. Front. Microbiol. 2019, 10, 1139.
- Kong, Q.; Stockinger, M.P.; Chang, Y.; Tashiro, H.; Lin, C.-L.G. The presence of rRNA sequences in polyadenylated RNA and its potential functions. Biotechnol. J. 2008, 3, 1041–1046.
- Mauro, V.P.; Edelman, G.M. rRNA-like sequences occur in diverse primary transcripts: Implications for the control of gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 422–427.
- Pánek, J.; Kolář, M.; Vohradský, J.; Valášek, L.S. An evolutionary conserved pattern of 18S rRNA sequence complementarity to mRNA 5′ UTRs and its implications for eukaryotic gene translation regulation. Nucleic Acids Res. 2013, 41, 7625–7634.
- Fujimura, T.; Esteban, R. Cap-snatching mechanism in yeast L-A double-stranded RNA virus. Proc. Natl. Acad. Sci. USA 2011, 108, 17667–17671.