Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Justin Matheson.
The endocannabinoid system (ECS) plays an integral role in maintaining metabolic homeostasis and may affect hunger, caloric intake, and nutrient absorption. Obesity has been associated with higher levels of the endogenous cannabinoid transmitters (endocannabinoids). Therefore, the ECS is an important target in obesity treatment. Modulating the enzymes that synthesize and degrade endocannabinoids, namely fatty acid amide hydrolase (FAAH), monoacylglycerol lipase (MAGL), and diacylglycerol lipase (DAGL), may be a promising strategy to treat obesity.
- obesity
- endocannabinoid system
- FAAH
- MAGL
- DAGL
1. Introduction
Obesity is a significant and ever-growing public health concern defined by a body mass index (BMI) of greater than 30 kg/m2. Global data indicate that age-standardized obesity rates rose from 3.2% in 1975 to 10.8% in 2014 for men and from 6.4% in 1975 to 14.9% in 2014 for women [1]. The latest data from the NCD Risk Factor Collaboration showed that in 2016, almost 2 billion adults (39% of the global adult population) were estimated to be overweight (BMI ≥ 25 kg/m2) and 671 million (12% of the global adult population) had obesity [2]. Obesity has been associated with numerous adverse health outcomes, including increased incidence of type II diabetes, cancers, and cardiovascular disease [3], and has both direct (medical) and indirect (nonmedical) costs that pose a significant global economic burden [4]. Developing effective therapeutic strategies to treat obesity is thus a global health priority.
The endocannabinoid system (ECS) was recognized as a potential target for obesity treatment in the early 2000’s [5]. The ECS is an evolutionarily conserved lipid signalling system that has widespread involvement in nearly all physiological processes, including energy homeostasis, synaptic plasticity, and feeding behaviours [6]. The major components of the ECS include: two canonical G-protein coupled receptors, the cannabinoid type-1 (CB1) and type-2 (CB2) receptors; the endogenous ligands (endocannabinoids), which are phospholipid derivatives containing a poly-unsaturated fatty acid moiety and a polar head group, either ethanolamine in the case of arachidonoylethanolamide (anandamide; AEA) or glycerol in the case of 2-arachidonoylglycerol (2-AG); and the enzymes responsible for the synthesis and degradation of the endocannabinoids [7]. One major synthetic pathway for 2-AG involves the enzyme diacylglycerol lipase (DAGL), while AEA synthesis is more varied [6]. DAGL has two isoforms, DAGLα and DAGLβ, which are encoded by two different human genes, DAGLA and DAGLB [8]. In terms of degradation, fatty acid amide hydrolase (FAAH), encoded by the human FAAH gene, is one of the most studied ECS enzymes, as it is responsible for the majority of AEA metabolism and is involved to a lesser extent in 2-AG metabolism [7]. Similarly, monoacylglycerol lipase (MAGL), encoded by the human MGLL gene, is responsible for the majority of 2-AG metabolism [7]. See Figure 1 for an overview of the ECS.
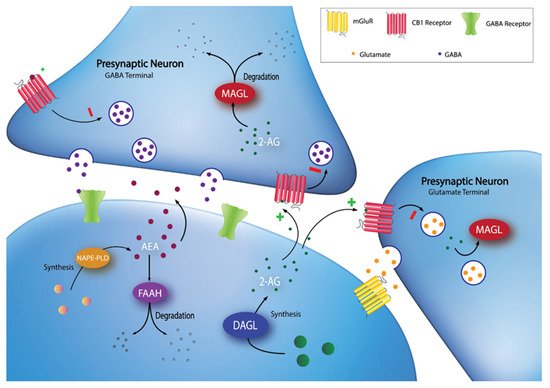
Figure 1. Synaptic localization of endocannabinoid synthesis and degradation. 2-AG is synthesized on-demand in the postsynaptic neuron by DAGL. Following synthesis, 2-AG diffuses into the synaptic cleft and activates CB1 receptors at GABA and glutamate terminals. 2-AG signaling can be terminated by MAGL degradation in the presynaptic terminal. AEA can be synthesized through multiple pathways (e.g., through a pathway involving NAPE-PLD), then diffuses into the synapse to activate CB1 receptors. Extracellular AEA undergoes reuptake into the postsynaptic cell and is hydrolyzed by FAAH.
The ECS has a multi-faceted role in the control of food intake and body weight, and it acts through both peripheral and central mechanisms. In the periphery, activation of CB1 receptors (e.g., by binding of AEA or 2-AG) promotes fat storage in adipocytes and increases lipogenesis, increases glucose uptake, and reduces satiety signals [9]. Centrally, activation of the ECS interferes with the control of hunger and satiety in multiple brain regions, including the hypothalamus and brain stem [9]. This likely involves a complex interplay between endocannabinoids and other neurotransmitters or hormones involved in hunger and satiety such as cholecystokinin (CCK), glucagon-like peptide 1 (GLP-1), and neuropeptide Y (NPY) [9]. Since the primary physiological function of the ECS is thought to shift energy balance towards energy storage, overactivation of the ECS likely contributes to obesity [5]. Numerous studies have found a positive association between circulating levels of endocannabinoids (AEA and/or 2-AG) and obesity in humans [10,11,12][10][11][12]. The elevated levels of endocannabinoids in human obesity are related to diet [13] and have been found to decrease after bariatric surgery [14].
Given that activation of the ECS increases food intake and energy storage, and that excessive activation of the ECS contributes to the pathogenesis of obesity, antagonism of the ECS has been explored as a potential therapeutic strategy [5,15][5][15]. In the early to mid-2000’s, the multi-center Rimonabant in Obesity (RIO) trials found significant evidence that rimonabant, a CB1 receptor antagonist/inverse agonist, reduced body weight in adults with obesity, and improved other secondary measures such as dyslipidemia and cardiometabolic risk factors [16,17][16][17]. Unfortunately, the promise of rimonabant as an obesity treatment was short-lived, as it was withdrawn from the market due to mounting evidence of serious psychiatric adverse effects, including increased risk of suicidal ideation and depression [18,19,20][18][19][20]. The promise of rimonabant to treat obesity led to efforts to develop and test numerous other CB1 receptor ligands that might have potential to modulate body weight and treat obesity. Interested readers are directed to our previous review of CB1 receptor ligands and their potential to treat obesity [15]. In brief, some potential strategies to target the CB1 receptor without producing serious psychiatric adverse effects include peripherally-restricted CB1 inverse agonists, CB1 receptor partial agonists, and CB1 receptor neutral antagonists.
Rather than directly targeting the CB1 receptor, another potential strategy to modulate ECS activity is to target the enzymes responsible for synthesis and/or degradation of the endocannabinoids. Two approaches can be undertaken: (1) targeting endocannabinoid degradation, i.e., by inhibiting FAAH or MAGL, and (2) targeting endocannabinoid biosynthesis, i.e., by inhibiting DAGL or another synthetic enzyme [8]. Given that obesity is associated with enhanced endocannabinoid tone, inhibiting endocannabinoid biosynthesis (e.g., with a DAGL inhibitor) is the most biologically plausible approach. However, the activity of ECS enzymes is complex, so it is hard to predict the effect of inhibiting ECS enzymes. For example, in some tissues FAAH inhibition may target 2-AG in addition to AEA, and it can both increase and decrease 2-AG concentrations [21]. There are also other substrates of FAAH that have a role in metabolism and satiety such as oleoylethanolamine (OEA) and palmitoylethanoamine (PEA). OEA is notable in particular as it has been shown to reduce food intake and suppress appetite, which is opposite to the effects of the endocannabinoids [22]. As FAAH metabolizes both pro-appetitive and anti-appetitive lipids, inhibition of FAAH would be expected to have mixed effects on appetite and body weight. It should be also noted that AEA and 2-AG have complex regulations; for example, AEA has been shown to inhibit metabolism and actions of 2-AG in the striatum [23].
2. Current Evidence for a Role of Modulating FAAH, MAGL, and DAGL Activity in Obesity-Related Outcomes
2.1. Animal Studies Involving Pharmacological Manipulation
Balsevich et al. (2018) investigated the role of FAAH in hypophagic responses to leptin [26][24]. Following a period of food deprivation, leptin administration induced expected reductions in body weight gain and food intake in saline-treated mice. Animals pre-treated with the FAAH inhibitor URB597 did not show these satiety responses following leptin treatment, suggesting that FAAH inhibition suppresses the effects of leptin on body weight and food intake. In another study, pharmacological inhibition of FAAH with PF-3845 had no effect on energy storage in rats [27][25]. Following exposure to a high-fat diet, PF-3845 and saline-treated animals showed similar decreases in body weight and food intake.
Stearoyl-CoA desaturase-1 (SCD1) is an enzyme involved in the synthesis of endogenous monounsaturated fatty acids (MUFAs). Due to its role in obesity, Liu et al. (2013) investigated functional interactions between SCD1 and ECS activity and found that MUFAs act as endogenous inhibitors of FAAH [28][26]. Under a high-fat diet, SCD1 knockout mice remained insulin-sensitive and glucose-tolerant compared to wildtype mice. When SCD1-deficient mice were treated with the FAAH inhibitor URB597, they became insulin resistant and showed increased sensitivity to glucose. Endogenous FAAH inhibition resulting from the SCD1-mediated production of MUFAs may thus contribute to the hyperinsulinemic phenotype in response to high-fat diets.
Two studies also investigated the use of DAGL inhibitors with obesity-related outcomes. Bisogno et al. (2013) found that the DAGLα inhibitor O-7460 significantly and dose-dependently reduced food intake in male mice under a high-fat diet [29][27]. The highest dose of O-7460 (12 mg/kg) was also associated with a small yet significant decrease in body weight. Palma-Chavez et al. (2019) used a shRNA DAGLα-inhibiting adenovirus to target tancytes of the hypothalamus and observed expression patterns of orexigenic (NPY) and anorexigenic (POMC) neuropeptides [30][28]. In fasting conditions, DAGLα inhibition was associated with reduced orexigenic and increased anorexigenic neuropeptides. Following glucose administration, DAGLα inhibition increased orexigenic and decreased anorexigenic neuropeptide expression. Control rats showed opposite responses in both conditions. Taken together, these findings indicate that DAGLα may regulate feeding behavior through the modulation of tancytic neuropeptides. DAGLα inhibition may reduce feeding through resulting decreases in 2-AG.
2.2. Animal Studies Involving Genetic Manipulation
The genetic deletion of Faah consistently produced obesity-related phenotypes across studies, as expected from resulting increases in AEA signaling. Greater body weight, fat mass, and triglyceride levels were observed in Faah knockout mice compared to wildtype under both standard and high-fat diets [31,32][29][30]. Faah-deficient mice also showed hepatic insulin resistance, which was associated with elevated liver triglyceride and diacylglycerol content [33][31]. The obesity phenotype found by Vaitheesvaran et al. (2012) in Faah-deficient mice was accompanied with greater total food intake under a regular diet [32][30]. In contrast, Touriño et al. (2010) found no effect of Faah deletion on food intake under either a standard or a high-fat diet [31][29]. However, Faah knockout mice showed greater reinforcement and motivational responses to food.
The FAAH C385A polymorphism is a common loss-of-function mutation in humans. The A-allele results in lower FAAH expression and has been associated with heightened obesity risk. Balsevich et al. (2018) generated a knock-in model of this variant and found impaired leptin sensitivity in A385A mice [26][24]. The expected reductions in food intake and body weight following leptin treatment seen in wildtype were absent in transgenic mice, suggesting a role of FAAH in leptin satiety responses.
In addition to hydrolyzing N-acylethanolamines (NAEs) such as AEA, FAAH terminates the signaling activity of N-acyl taurines (NATs). Grevengoed et al. (2019) engineered a Faah knock-in model that selectively impaired NAT catabolism without affecting NAE degradation [34][32]. The S268D substitution did not replicate the obesity phenotypes found in the other studies. Instead, transgenic mice showed an improved metabolic profile compared to wildtypes. These findings confirm that obesity phenotypes found in Faah-deficient models are the result of endocannabinoid accumulation rather than increased NATs. Elevated NAT levels may instead have beneficial effects on energy storage and metabolism, which remain to be investigated.
Five studies investigated the role of Mgll in energy storage and obesity-related measures. Overexpression of Mgll in forebrain neurons lowered endocannabinoid levels, causing expected reductions in weight gain and adiposity [35][33]. Interestingly, intestinal overexpression of Mgll produced the opposite phenotype, characterized by increased body fat mass and weight gain [36][34]. These contradicting findings may reflect tissue-specific functions of MAGL. Similarly, under an obesogenic diet, the genetic deletion of Mgll was associated with paradoxical improvements in metabolic function. Compared to wildtypes, Mgll-deficient mice experienced less weight gain, had lower plasma triglyceride levels, and lower hepatic triglyceride content [37,38][35][36]. Insulin sensitivity and glucose tolerance were also improved [38][36]. Under a low-fat diet, knockout mice had lower fat mass and body weight despite showing no significant differences in food intake [39][37]. Reduced triglyceride and cholesterol levels were also found in male Mgll knockout mice, under both low and high-fat diets. When males and females were considered together, this effect was only found in response to a high-fat diet. The potential sex-specific effects of Mgll function on energy storage were not investigated by the other studies, as they included only male mice.
Our search identified only one genetic manipulation study on Dagla and obesity-related measures. Powell et al. (2015) found that Dagla deletion improved metabolic function [40][38]. Compared to wildtype, knockout mice had reduced body weight, fat mass, total triglycerides and cholesterol. Food intake and fasting insulin levels were also reduced in Dagla-deficient mice.
2.3. Human Studies Involving Genetic Association
The majority of studies (26 out of 30) included one specific FAAH SNP, a missense mutation (C385A) associated with a change in amino acid sequence at position 129 from proline to threonine (Pro129Thr). This SNP was originally identified in 2002 and was found to be strongly associated with problematic substance use [41][39]. The Pro129Thr variant was found to have normal catalytic activity but an enhanced sensitivity to proteolytic degradation [41][39]. Subsequent functional characterization of the variant found that the mutant FAAH enzyme had less than half of the expression and activity of the wildtype enzyme [42][40].
Of the 26 studies included in the present review, 19 found a significant association between the C385A SNP and at least one obesity-related outcome [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59]. Following up on their initial discovery of the C385A SNP, Sipe and colleagues were the first to identify a significant association between the A allele and increased likelihood of being overweight or obese in a sample of 1688 White adults and 614 Black adults (but not in 365 Asian adults) [57][55]. In support of these findings, Monteleone et al. (2008) found that the A allele was significantly more common in women who were overweight or obese compared to healthy weight controls (n = 299) [55][53]. Yagin et al. (2019) similarly found the A allele to be more common in women who were overweight or obese (n = 180) compared to health weight controls (n = 86) [60][58]. Thethi et al. (2020) recently reported another significant association between the A allele and obesity, though the association was no longer significant when controlling for age, race, sex, waist-to-hip ratio, and cholesterol [58][56]. Further support for an association between the C385A variant and increased risk of obesity comes from studies including continuous outcomes. For example, the A allele was associated with higher BMI, fat mass, and waist circumference in a sample of 70 participants with Type-II diabetes mellitus who were obese [50][48]. Yagin et al. (2019) also found a significant association between the A allele and higher BMI, waist circumference, neck circumference, waist-to-height ratio, and body fat mass [60][58], while Zhang et al. (2009) found an association between the A allele and higher BMI and triglyceride levels in a sample of 1644 participants from 261 pedigrees [61][59]. Further, Vazquez-Roque et al. (2011) found that A-allele carriers had a significantly greater maximum tolerated volume after a nutrient drink test, indicating lower satiety [59][57].
In contrast, multiple studies have failed to replicate the association between the A allele and obesity. Seven studies found no significant association between the SNP and any obesity-related outcomes [62,63,64,65,66,67,68][60][61][62][63][64][65][66]. A few studies actually found the reverse association—either a significant association between the wildtype (C/C) genotype and obesity [53,56][51][54] or an association of the wildtype (C/C) genotype with worse cardiometabolic outcomes, e.g., higher triglycerides and metabolic biomarkers [46][44]. Studies conducted by De Luis and colleagues failed to find any significant association between the C385A SNP and a wide range of obesity-related outcomes including anthropometric outcomes (BMI, body weight, waist circumference, etc.) and circulating lipids [44,45,46,47,48,49,51,52][42][43][44][45][46][47][49][50].
Interestingly, a number of studies have suggested that while the C385A SNP may not directly impact obesity-related outcomes at baseline, there may be an impact of the variant on changes in outcomes following either surgical or lifestyle interventions for obesity. Aberle et al. (2007) found that A-allele carriers had greater decreases in triglyceride and cholesterol levels after a 6-week low-fat diet intervention (n = 451 obese and dyslipidemic participants) [43][41]. Similarly, De Luis et al. (2011) found that A-allele carriers had greater improvement in a range of metabolic outcomes after 3 months of a hypocaloric dietary intervention (n = 122) [47][45], while De Luis et al. (2010) found greater weight loss in A-allele carriers at 9 and 12 months after biliopancreatic diversion surgery (n = 67) [51][49]. In contrast, three other studies by De Luis and colleagues found that the A-allele was associated with worse outcomes following either a low-fat or low-carbohydrate diet intervention for 3 months (n = 248) [52][50], and also following a 3-month enriched monounsaturated fat hypocaloric diet (n = 95) [44][42] and a 3-month enriched polyunsaturated fat hypocaloric diet (n = 99) [49][47]. Finally, Knoll et al. (2012) failed to show any association of the C385A variant and obesity-related outcomes after a 1-year diet and exercise intervention in a group of 453 overweight and obese children and adolescents [63][61].
In addition to examining associations between the FAAH C385A variant and obesity-related outcomes, four studies examined associations with other FAAH SNPs [53,56,61,64][51][54][59][62]. In a large sample of 5109 French Caucasian participants, Durand et al. (2008) failed to find an association between 10 FAAH SNPs and childhood obesity or Type-II diabetes mellitus, but did find a nominal association between 5 FAAH SNPs (rs6429600, rs324419, rs324418, rs2295633, and rs7520850) and Class III adult obesity [53][51]. Lieb et al. (2009) failed to find an association between 9 FAAH SNPs and obesity-related outcomes in a sample of 2415 participants from a longitudinal cohort [64][62]. Muller et al. (2010) found some additional evidence of association between FAAH gene variants and obesity—the G allele of rs2295632 was associated with early-onset obesity in 521 trios (children/adolescents and both biological parents), though this finding did not replicate in a second cohort of trios, and there was no association with obesity in adults (n = 8491 participants from a population-based study sample) [56][54]. Finally, Zhang et al. (2009) did not find an association between four FAAH SNPs (rs324418, rs1984490, rs2145408 and rs4141964) and any obesity-related traits [61][59].
Three studies used bioinformatic approaches to identify novel genetic variants in FAAH and MGLL genes associated with obesity or obesity-related traits [69,70,71][67][68][69]. Bhatia et al. (2010) used data from a large clinical trial cohort (n = 289) to identify rare variants in FAAH and MGLL associated with obesity, and found one significantly associated region of approximately 5 Kbp in the upstream regulatory region of each gene [69][67]. Harismendy et al. (2010) similarly used data from the same clinical trial cohort to identify one interval in the FAAH promoter and three intervals in MGLL (in the promoter, intron 2, and intron 3) that were all significantly associated with obesity [70][68]. Kuk et al. (2013) identified three potentially causal rare variants in MGLL associated with obesity, while also finding an interaction between two rare variants in FAAH that may increase the risk of obesity [71][69].
References
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396.
- NCD Risk Factor Collaboration. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128,9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642.
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88.
- Spieker, E.A.; Pyzocha, N. Economic Impact of Obesity. Prim. Care 2016, 43, 83–95.
- Richey, J.M.; Woolcott, O. Re-visiting the Endocannabinoid System and Its Therapeutic Potential in Obesity and Associated Diseases. Curr. Diab. Rep. 2017, 17, 99.
- Lu, H.C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525.
- Pamplona, F.A.; Takahashi, R.N. Psychopharmacology of the endocannabinoids: Far beyond anandamide. J. Psychopharmacol. 2012, 26, 7–22.
- Gregus, A.M.; Buczynski, M.W. Druggable Targets in Endocannabinoid Signaling. Adv. Exp. Med. Biol. 2020, 1274, 177–201.
- Schulz, P.; Hryhorowicz, S.; Rychter, A.M.; Zawada, A.; Słomski, R.; Dobrowolska, A.; Krela-Kaźmierczak, I. What Role Does the Endocannabinoid System Play in the Pathogenesis of Obesity? Nutrients 2021, 13, 373.
- Rahmanian, M.; Lotfi Yaghin, N.; Alizadeh, M. Blood Level of 2-arachidonoyl glycerol (2-AG), Neuropeptide Y and Omentin and Their Correlation with Food Habits in Obese Women. Galen. Med. J. 2020, 9, e1721.
- Côté, M.; Matias, I.; Lemieux, I.; Petrosino, S.; Alméras, N.; Després, J.P.; Di Marzo, V. Circulating endocannabinoid levels, abdominal adiposity and related cardiometabolic risk factors in obese men. Int. J. Obes. 2007, 31, 692–699.
- Matias, I.; Gatta-Cherifi, B.; Tabarin, A.; Clark, S.; Leste-Lasserre, T.; Marsicano, G.; Piazza, P.V.; Cota, D. Endocannabinoids measurement in human saliva as potential biomarker of obesity. PLoS ONE 2012, 7, e42399.
- Yagin, N.L.; Hajjarzadeh, S.; Aliasgharzadeh, S.; Aliasgari, F.; Mahdavi, R. The association of dietary patterns with endocannabinoids levels in overweight and obese women. Lipids Health Dis. 2020, 19, 161.
- Azar, S.; Sherf-Dagan, S.; Nemirovski, A.; Webb, M.; Raziel, A.; Keidar, A.; Goitein, D.; Sakran, N.; Shibolet, O.; Tam, J.; et al. Circulating Endocannabinoids Are Reduced Following Bariatric Surgery and Associated with Improved Metabolic Homeostasis in Humans. Obes. Surg. 2019, 29, 268–276.
- Murphy, T.; Le Foll, B. Targeting the Endocannabinoid CB1 Receptor to Treat Body Weight Disorders: A Preclinical and Clinical Review of the Therapeutic Potential of Past and Present CB1 Drugs. Biomolecules 2020, 10, 855.
- Christopoulou, F.D.; Kiortsis, D.N. An overview of the metabolic effects of rimonabant in randomized controlled trials: Potential for other cannabinoid 1 receptor blockers in obesity. J. Clin. Pharm. Ther. 2011, 36, 10–18.
- Leite, C.E.; Mocelin, C.A.; Petersen, G.O.; Leal, M.B.; Thiesen, F.V. Rimonabant: An antagonist drug of the endocannabinoid system for the treatment of obesity. Pharmacol. Rep. 2009, 61, 217–224.
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713.
- Moreira, F.A.; Crippa, J.A. The psychiatric side-effects of rimonabant. Braz. J. Psychiatry 2009, 31, 145–153.
- Le Foll, B.; Gorelick, D.A.; Goldberg, S.R. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology 2009, 205, 171–174.
- Toczek, M.; Malinowska, B. Enhanced endocannabinoid tone as a potential target of pharmacotherapy. Life Sci. 2018, 204, 20–45.
- Laleh, P.; Yaser, K.; Alireza, O. Oleoylethanolamide: A novel pharmaceutical agent in the management of obesity-an updated review. J. Cell Physiol. 2019, 234, 7893–7902.
- Maccarrone, M.; Rossi, S.; Bari, M.; De Chiara, V.; Fezza, F.; Musella, A.; Gasperi, V.; Prosperetti, C.; Bernardi, G.; Finazzi-Agrò, A.; et al. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat. Neurosci. 2008, 11, 152–159.
- Balsevich, G.; Sticht, M.; Bowles, N.P.; Singh, A.; Lee, T.T.Y.; Li, Z.; Chelikani, P.K.; Lee, F.S.; Borgland, S.L.; Hillard, C.J.; et al. Role for fatty acid amide hydrolase (FAAH) in the leptin-mediated effects on feeding and energy balance. Proc. Natl. Acad. Sci. USA 2018, 115, 7605–7610.
- Cifani, C.; Avagliano, C.; Micioni Di Bonaventura, E.; Giusepponi, M.E.; De Caro, C.; Cristiano, C.; La Rana, G.; Botticelli, L.; Romano, A.; Calignano, A.; et al. Modulation of Pain Sensitivity by Chronic Consumption of Highly Palatable Food Followed by Abstinence: Emerging Role of Fatty Acid Amide Hydrolase. Front. Pharmacol. 2020, 11, 266.
- Liu, J.; Cinar, R.; Xiong, K.; Godlewski, G.; Jourdan, T.; Lin, Y.; Ntambi, J.M.; Kunos, G. Monounsaturated fatty acids generated via stearoyl CoA desaturase-1 are endogenous inhibitors of fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2013, 110, 18832–18837.
- Bisogno, T.; Mahadevan, A.; Coccurello, R.; Chang, J.W.; Allarà, M.; Chen, Y.; Giacovazzo, G.; Lichtman, A.; Cravatt, B.; Moles, A.; et al. A novel fluorophosphonate inhibitor of the biosynthesis of the endocannabinoid 2-arachidonoylglycerol with potential anti-obesity effects. Br. J. Pharmacol. 2013, 169, 784–793.
- Palma-Chavez, A.; Konar-Nié, M.; Órdenes, P.; Maurelia, F.; Elizondo-Vega, R.; Oyarce, K.; López, S.; Rojas, J.; Steinberg, X.; García-Robles, M.A.; et al. Glucose Increase DAGLα Levels in Tanycytes and Its Inhibition Alters Orexigenic and Anorexigenic Neuropeptides Expression in Response to Glucose. Front. Endocrinol. 2019, 10, 647.
- Touriño, C.; Oveisi, F.; Lockney, J.; Piomelli, D.; Maldonado, R. FAAH deficiency promotes energy storage and enhances the motivation for food. Int. J. Obes. 2010, 34, 557–568.
- Vaitheesvaran, B.; Yang, L.; Hartil, K.; Glaser, S.; Yazulla, S.; Bruce, J.E.; Kurland, I.J. Peripheral Effects of FAAH Deficiency on Fuel and Energy Homeostasis: Role of Dysregulated Lysine Acetylation. PLoS ONE 2012, 7, e33717.
- Brown, W.H.; Gillum, M.P.; Lee, H.-Y.; Camporez, J.P.G.; Zhang, X.-M.; Jeong, J.K. Fatty acid amide hydrolase ablation promotes ectopic lipid storage and insulin resistance due to centrally mediated hypothyroidism. Proc. Natl. Acad. Sci. USA 2012, 109, 14966–14971.
- Grevengoed, T.J.; Trammell, S.A.J.; McKinney, M.K.; Petersen, N.; Cardone, R.L.; Svenningsen, J.S.; Ogasawara, D.; Nexøe-Larsen, C.C.; Knop, F.K.; Schwartz, T.W.; et al. N-acyl taurines are endogenous lipid messengers that improve glucose homeostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 24770–24778.
- Jung, K.-M.; Clapper, J.R.; Fu, J.; D’Agostino, G.; Guijarro, A.; Thongkham, D.; Avanesian, A.; Astarita, G.; DiPatrizio, N.V.; Frontini, A.; et al. 2-Arachidonoylglycerol Signaling in Forebrain Regulates Systemic Energy Metabolism. Cell Metab. 2012, 15, 299–310.
- Chon, S.-H.; Douglass, J.D.; Zhou, Y.X.; Malik, N.; Dixon, J.L.; Brinker, A.; Quadro, L.; Storch, J. Over-Expression of Monoacylglycerol Lipase (MGL) in Small Intestine Alters Endocannabinoid Levels and Whole Body Energy Balance, Resulting in Obesity. PLoS ONE 2012, 7, e43962.
- Tardelli, M.; Bruschi, F.V.; Claudel, T.; Fuchs, C.D.; Auer, N.; Kunczer, V.; Stojakovic, T.; Scharnagl, H.; Habib, A.; Grabner, G.F.; et al. Lack of monoacylglycerol lipase prevents hepatic steatosis by favoring lipid storage in adipose tissue and intestinal malabsorption. J. Lipid Res. 2019, 60, 1284–1292.
- Yoshida, K.; Kita, Y.; Tokuoka, S.M.; Hamano, F.; Yamazaki, M.; Sakimura, K.; Kano, M.; Shimizu, T. Monoacylglycerol lipase deficiency affects diet-induced obesity, fat absorption, and feeding behavior in CB1 cannabinoid receptor-deficient mice. FASEB J. 2019, 33, 2484–2497.
- Douglass, J.D.; Zhou, Y.X.; Wu, A.; Zadrogra, J.A.; Gajda, A.M.; Lackey, A.I.; Lang, W.; Chevalier, K.M.; Sutton, S.W.; Zhang, S.P.; et al. Global deletion of MGL in mice delays lipid absorption and alters energy homeostasis and diet-induced obesity. J. Lipid Res. 2015, 56, 1153–1171.
- Powell, D.R.; Gay, J.P.; Wilganowski, N.; Doree, D.; Savelieva, K.V.; Lanthorn, T.H.; Read, R.; Vogel, P.; Hansen, G.M.; Brommage, R.; et al. Diacylglycerol Lipase α Knockout Mice Demonstrate Metabolic and Behavioral Phenotypes Similar to Those of Cannabinoid Receptor 1 Knockout Mice. Front. Endocrinol. 2015, 6, 86.
- Sipe, J.C.; Chiang, K.; Gerber, A.L.; Beutler, E.; Cravatt, B.F. A missense mutation in human fatty acid amide hydrolase associated with problem drug use. Proc. Natl. Acad. Sci. USA 2002, 99, 8394–8399.
- Chiang, K.P.; Gerber, A.L.; Sipe, J.C.; Cravatt, B.F. Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: Evidence for a link between defects in the endocannabinoid system and problem drug use. Hum. Mol. Genet. 2004, 13, 2113–2119.
- Aberle, J.; Fedderwitz, I.; Klages, N.; George, E.; Beil, F.U. Genetic Variation in Two Proteins of the Endocannabinoid System and their Influence on Body Mass Index and Metabolism under Low Fat Diet. Horm. Metab. Res. 2007, 39, 395–397.
- de Luis, D.; Aller, R.; Izaola, O.; Conde, R.; de la Fuente, B.; Sagrado, M.G. Genetic variation in the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) and their influence on weight loss and insulin resistance under a high monounsaturated fat hypocaloric diet. J. Diabetes Complicat. 2013, 27, 235–239.
- de Luis, D.A.; Aller, R.; Izaola, O.; Conde, R.; Sagrado, M.G.; Primo, D.; Castro, M.J. Relationship among metabolic syndrome, C358A polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) and insulin resistance. J. Diabetes Complicat. 2012, 26, 328–332.
- de Luis, D.A.; González Sagrado, M.; Aller, R.; Izaola, O.; Conde, R. Relation of C358A polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) with obesity and insulin resistance. Nutr. Hosp. 2010, 25, 993–998.
- de Luis, D.A.; Gonzalez Sagrado, M.; Aller, R.; Izaola, O.; Conde, R. Effects of C358A missense polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase on weight loss after a hypocaloric diet. Metab. Clin. Exp. 2011, 60, 730–734.
- de Luis, D.A.; Gonzalez Sagrado, M.; Aller, R.; Izaola, O.; Conde, R.; Romero, E. C358A missense polymorphism of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) and visfatin levels in obese females. Int. J. Obes. 2010, 34, 511–515.
- de Luis, D.A.; Izaola, O.; Aller, R.; de La Fuente, B.; Pacheco, D. Effects of C358A polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) on weight loss, adipocytokines levels, and insulin resistance after a high polyunsaturated fat diet in obese patients. J. Endocrinol. Investig. 2013, 36, 965–969.
- de Luis, D.A.; Sagrado, M.G.; Aller, R.; Izaola, O.; Conde, R.; Romero, E. C358A missense polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) and insulin resistance in patients with diabetes mellitus type 2. Diabetes Res. Clin. Pract. 2010, 88, 76–80.
- de Luis, D.A.; Sagrado, M.G.; Pacheco, D.; Terroba, M.C.; Martin, T.; Cuellar, L.; Ventosa, M. Effects of C358A missense polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase on weight loss and cardiovascular risk factors 1 year after biliopancreatic diversion surgery. Surg. Obes. Relat. Dis. 2010, 6, 516–520.
- de Luis, D.A.; Sagrado, M.G.; Aller, R.; Izaola, O.; Conde, R. Effects of C358A missense polymorphism of the degrading enzyme fatty acid amide hydrolase on weight loss, adipocytokines, and insulin resistance after 2 hypocaloric diets. Metab. Clin. Exp. 2010, 59, 1387–1392.
- Durand, E.; Lecoeur, C.; Delplanque, J.; Benzinou, M.; Degraeve, F.; Boutin, P.; Marre, M.; Balkau, B.; Charpentier, G.; Froguel, P.; et al. Evaluating the Association of FAAH Common Gene Variation with Childhood, Adult Severe Obesity and Type 2 Diabetes in the French Population. Obes. Facts 2008, 1, 305–309.
- Grolmusz, V.K.; Stenczer, B.; Fekete, T.; Szendei, G.; Patócs, A.; Rácz, K.; Reismann, P. Lack of Association between C385A Functional Polymorphism of the Fatty Acid Amide Hydrolase Gene and Polycystic Ovary Syndrome. Exp. Clin. Endocrinol. Diabetes 2013, 121, 338–342.
- Monteleone, P.; Tortorella, A.; Martiadis, V.; Di Filippo, C.; Canestrelli, B.; Maj, M. The cDNA 385C to A missense polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) is associated with overweight/obesity but not with binge eating disorder in overweight/obese women. Psychoneuroendocrinology 2008, 33, 546–550.
- Müller, T.D.; Brönner, G.; Wandolski, M.; Carrie, J.; Nguyen, T.T.; Greene, B.H.; Scherag, A.; Grallert, H.; Vogel, C.I.; Scherag, S.; et al. Mutation screen and association studies for the fatty acid amide hydrolase (FAAH) gene and early onset and adult obesity. BMC Med. Genet. 2010, 11, 2.
- Sipe, J.C.; Waalen, J.; Gerber, A.; Beutler, E. Overweight and obesity associated with a missense polymorphism in fatty acid amide hydrolase (FAAH). Int. J. Obes. 2005, 29, 755–759.
- Thethi, T.K.; Sigel, A.; Japa, S.; Katalenich, B.; Liu, S.; Nguyen, T.; Larrazolo, J.; Syu, S.; Carefoot, E.; McDuffie, R.; et al. Racial and sex differences in the polymorphisms of the endocannabinoid receptor genes in obesity. J. Diabetes Complicat. 2020, 34, 107682.
- Vazquez-Roque, M.I.; Camilleri, M.; Vella, A.; Carlson, P.; Laugen, J.; Zinsmeister, A.R. Association of genetic variation in cannabinoid mechanisms and gastric motor functions and satiation in overweight and obesity. Neurogastroenterol. Motil. 2011, 23, 637-e257.
- Yagin, N.L.; Aliasgari, F.; Aliasgharzadeh, S.; Mahdavi, R.; Akbarzadeh, M. The influence of the fatty acid amide hydrolase 385C>A single nucleotide polymorphisms on obesity susceptibility. Mol. Biol. Rep. 2019, 46, 5049–5055.
- Zhang, Y.; Sonnenberg, G.E.; Baye, T.M.; Littrell, J.; Gunnell, J.; DeLaForest, A.; MacKinney, E.; Hillard, C.J.; Kissebah, A.H.; Olivier, M.; et al. Obesity-related dyslipidemia associated with FAAH, independent of insulin response, in multigenerational families of Northern European descent. Pharmacogenomics 2009, 10, 1929–1939.
- Jensen, D.P.; Andreasen, C.H.; Andersen, M.K.; Hansen, L.; Eiberg, H.; Borch-Johnsen, K.; Jørgensen, T.; Hansen, T.; Pedersen, O. The functional Pro129Thr variant of the FAAH gene is not associated with various fat accumulation phenotypes in a population-based cohort of 5801 whites. J. Mol. Med. 2007, 85, 445–449.
- Knoll, N.; Volckmar, A.L.; Pütter, C.; Scherag, A.; Kleber, M.; Hebebrand, J.; Hinney, A.; Reinehr, T. The Fatty Acid Amide Hydrolase (FAAH) Gene Variant rs324420 AA/AC is not Associated with Weight Loss in a 1-Year Lifestyle Intervention for Obese Children and Adolescents. Horm. Metab. Res. 2012, 44, 75–77.
- Lieb, W.; Manning, A.K.; Florez, J.C.; Dupuis, J.; Cupples, L.A.; McAteer, J.B.; Vasan, R.S.; Hoffmann, U.; O’Donnell, C.J.; Meigs, J.B.; et al. Variants in the CNR1 and the FAAH Genes and Adiposity Traits in the Community. Obesity 2009, 17, 755–760.
- Mansouri, E.; Nobrega, J.N.; Hill, M.N.; Tyndale, R.F.; Lee, F.S.; Hendershot, C.S.; Best, L.M.; Di Ciano, P.; Balsevich, G.; Sloan, M.E.; et al. D3 dopamine receptors and a missense mutation of fatty acid amide hydrolase linked in mouse and men: Implication for addiction. Neuropsychopharmacology 2020, 45, 745–752.
- Martins, C.J.D.M.; Genelhu, V.; Pimentel, M.M.G.; Celoria, B.M.J.; Mangia, R.F.; Aveta, T.; Silvestri, C.; Di Marzo, V.; Francischetti, E.A. Circulating Endocannabinoids and the Polymorphism 385C>A in Fatty Acid Amide Hydrolase (FAAH) Gene May Identify the Obesity Phenotype Related to Cardiometabolic Risk: A Study Conducted in a Brazilian Population of Complex Interethnic Admixture. PLoS ONE 2015, 10, e0142728.
- Papazoglou, D.; Panagopoulos, I.; Papanas, N.; Gioka, T.; Papadopoulos, T.; Papathanasiou, P.; Kaitozis, O.; Papatheodorou, K.; Maltezos, E. The Fatty Acid Amide Hydrolase (FAAH) Pro129Thr Polymorphism is not Associated with Severe Obesity in Greek Subjects. Horm. Metab. Res. 2008, 40, 907–910.
- Yagin, N.L.; Aliasgari, F.; Alizadeh, M.; Aliasgharzadeh, S.; Mahdavi, R. Comparison of endocannabinoids levels, FAAH gene polymorphisms, and appetite regulatory substances in women with and without binge eating disorder: A cross—Sectional study. Nutr. Res. 2020, 83, 86–93.
- Bhatia, G.; Bansal, V.; Harismendy, O.; Schork, N.J.; Topol, E.J.; Frazer, K.; Bafna, V. A Covering Method for Detecting Genetic Associations between Rare Variants and Common Phenotypes. PLOS Comput. Biol. 2010, 6, e1000954.
- Harismendy, O.; Bansal, V.; Bhatia, G.; Nakano, M.; Scott, M.; Wang, X.; Dib, C.; Turlotte, E.; Sipe, J.C.; Murray, S.S.; et al. Population sequencing of two endocannabinoid metabolic genes identifies rare and common regulatory variants associated with extreme obesity and metabolite level. Genome Biol. 2010, 11, R118.
- Kuk, A.Y.C.; Li, X.; Xu, J. A fast collapsed data method for estimating haplotype frequencies from pooled genotype data with applications to the study of rare variants. Stat. Med. 2013, 32, 1343–1360.
More