Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Robb Krumlauf and Version 2 by Camila Xu.
Hox genes play key roles in axial patterning and regulating the regional identity of cells and tissues in a wide variety of animals from invertebrates to vertebrates.
- Hox genes
- transcriptional regulation
- transcription factors
1. Introduction
Animals display remarkable variety in their body plans and there is great interest in understanding the degree to which conserved and distinct mechanisms underlie this diversity in the formation and elaboration of basic body plans in animal evolution. In chordate evolution, there is emerging evidence for a deeply conserved regulatory network, involving transcription factors (TFs) and signaling pathways, that governs patterning along the anterior–posterior (A–P) body axis [1][2][3][4][5][6][1,2,3,4,5,6]. Remarkably, despite very different morphologies among chordates, many key TFs and components of major signaling pathways (e.g., Wnts and FGFs), known to regulate developmental processes, have been shown to be similarly aligned along the A–P axis. This suggests that regulatory interactions between signaling pathways and core TFs set up a conserved gene regulatory network (GRN) that guides the formation of the basic body plan and patterning of the A–P axis. However, the question of how TFs are coupled to these ancient signaling pathways and how they integrate responses to signaling gradients is not fully understood.
The highly conserved HOX family of TFs are an example of TFs that are coupled to this ancient GRN. Hox genes are known to play key roles in axial patterning and regulating the regional identity of cells and tissues in a wide variety of animals from invertebrates to vertebrates [7][8][9][10][11][7,8,9,10,11]. The clustered Hox genes exhibit an interesting property known as collinearity [12][13][14][15][16][12,13,14,15,16]. Genes in the four mammalian Hox clusters are all transcribed in the same 5′ to 3′ direction with respect to transcription, and the order of Hox genes in each cluster on a chromosome corelates with their temporal and spatial expression domains and functions along the A–P axis of developing embryos (Figure 1). These nested domains of expression generate a combinatorial Hox code, which provides a molecular framework that serves as a key regulatory step in specifying regional identities and properties of tissues along the A–P axis. A wide variety of studies in different species and cell culture models have revealed that the nested domains of Hox expression along the A–P axis arise in part through the ability of Hox clusters to integrate and respond to opposing signaling gradients, such as those of Retinoic acid (RA), Fibroblast growth factors (Fgfs) and Wingless related integration sites (WNTs) [5][17][18][19][20][21][22][23][24][25][26][27][28][29][5,17,18,19,20,21,22,23,24,25,26,27,28,29]. Hence, it is important to understand the regulatory mechanisms through which signaling pathways are able to coordinately control the precise patterns of the transcription of the clustered Hox genes required for their roles in specifying diverse morphologic features along the A–P axis.
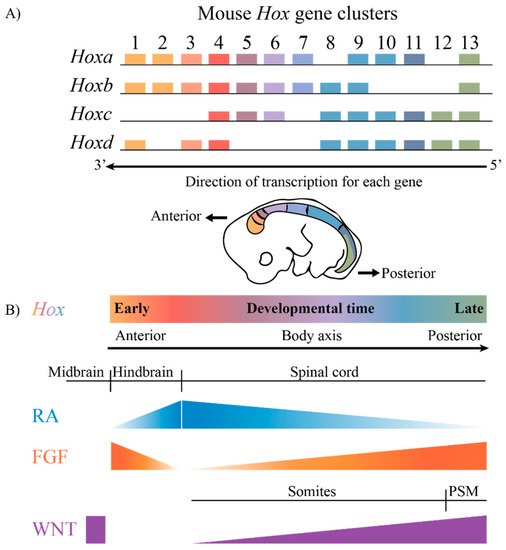
Figure 1. The mammalian Hox gene clusters and the conserved signaling pathways that play a role in defining the Hox gene expression profiles. (A) In mammals, there are four clusters of Hox genes, each on different chromosomes. They exhibit spatial and temporal collinearity, such that 3′ Hox genes are expressed early in development as well as more anteriorly in an embryo generating nested domains of expression as depicted in the drawing of an E10 mouse embryo. (B) The restricted domains of Hox expression arise through an integration of signaling molecules such as RA, FGF and WNT, which are expressed in gradients along the embryonic axis. PSM, presomitic mesoderm.
In the case of RA signaling, Hox genes are direct transcriptional targets of retinoids, and their response to RA signaling involves retinoic acid response elements (RAREs) embedded within and adjacent to the Hox clusters [18][30][31][32][18,30,31,32]. These RAREs are cis-regulatory components of RA-dependent enhancers that provide regulatory inputs both locally on adjacent Hox genes and over a long range to coordinately regulate multiple genes in a Hox cluster [33][34][35][36][37][38][33,34,35,36,37,38]. This tightly clustered organization of cis-regulatory elements and the Hox genes they control raises interesting questions with respect to roles for chromosome topology, epigenetic modifications, dynamics of transcription and the underlying transcriptional mechanisms for how enhancers display selectivity or competition between genes, and they may be shared by multiple genes in a cluster [39]. It is important that these diverse aspects of transcriptional regulation are properly coordinated to ensure the right spatial and temporal patterns and appropriate levels of expression needed for their roles in axial patterning.
The advent of new technologies for investigating the dynamics of interactions that underlie the activation of transcription are generating surprising findings. These observations challenge the widely postulated role of stable long-term enhancer promoter interactions and the notion of a single RNA polymerase with a small number of components regulating transcription [40][41][42][43][44][45][46][47][48][49][40,41,42,43,44,45,46,47,48,49]. New models suggest that dynamic condensates and mechanisms involving a series of rapid and complex interactions underlie the activation of transcription and the regulation of gene expression. It will be interesting and important to understand how this newly emerging picture of the dynamic molecular mechanisms governing transcription plays a role in modulating the inputs controlling the coordinated expression of the clustered Hox genes.
2. Regulatory Features
2.1. Enhancers
Enhancers were first discovered in simian virus 40 (SV40), where it was found that they function in an orientation-independent manner to stimulate transcription on heterologous genes [50]. Since then, a variety of analyses have revealed that animal genomes contain a large number of putative enhancers, out numbering coding genes [51][52][51,52]. It is challenging to identify cis-regulatory elements, such as enhancers, encoded in the genome through sequence analyses and computational methods alone [53]. Major efforts have been made to find ways of identifying and characterizing enhancers and their properties on a genome-wide and individual basis, which is important to facilitate our ability to decode regulatory information embedded in the genome [54][55][56][57][58][59][60][54,55,56,57,58,59,60]. While many development specific enhancers, including some of those discovered in the Hox clusters, are evolutionarily conserved [35][61][62][63][35,61,62,63], many adult or tissue-specific enhancers can be highly variable across species [64][65][64,65]. Even when enhancers are highly conserved, it can be challenging to understand the information content and the critical arrangements of the cis-elements that govern their ability to regulate expression [54][59][60][54,59,60]. Furthermore, highly conserved patterns of gene expression can arise through enhancers that display divergence [65]. Enhancers serve to stimulate transcription by integrating a variety of different regulatory inputs and binding sites for TFs to confer precise temporal, spatial and cell-type specific gene expression programs. Precise regulatory outputs from enhancers do not require that upstream factors have highly restricted domains of expression and can arise through the cumulative integration of weak, imprecise or wide-spread inputs by TFs [66][67][66,67]. The convergence of inputs can result in the integration of disparate and very broad patterns of regulatory signals into robust and tightly controlled specific outputs. Similarly, clusters of weak enhancers can synergize to serve as super enhancers to robustly regulate gene expression [68].
Enhancers can be located directly upstream of a gene or up to over a megabase away from its target gene promoter [69][70][69,70]. They frequently reside within introns of genes, even in ones they do not regulate, [70], and there is evidence for enhancers and cis-regulatory elements embedded in coding exons, including those of Hox genes [71][72][73][71,72,73]. Studies have shown that enhancer regions are themselves transcriptionally active. Several groups have demonstrated that non-coding enhancer RNAs are more than just transcriptional noise or byproducts of the transcriptional machinery, but are useful indicators in predicting active enhancers [74].
A challenge in identifying the targets of enhancer activity is that they can function independently of their orientation with respect to target genes and can make long-range enhancer–promoter contacts to more than the near adjacent genes [75]. Typically, an average vertebrate enhancer can be ~5 to 50 kb away from target promoters and ~1 to 10 kb away in the more compact Drosophila genome [39]. Intriguingly, the proximity of an enhancer to its target promoter required for functional activity is variable. Hence, there are no clear rules on how close enhancers should be positioned relative to the promoters they activate. Some studies have shown through proximity-dependent ligation techniques, such as 3C (Chromatin conformation capture), that enhancers physically come into contact with promoters, resulting in the activation of gene transcription [76]. Imaging approaches have shown that following the activation of genes, the distance between the enhancers and their target promoter tends to increase, suggesting a change in their interactions dependent upon their activity state [77]. This raises fundamental questions, such as, how do enhancers locate and distinguish between the target genes they activate; what confers enhancer–promoter specificity; and what degree of proximity is essential for the enhancer interactions required for gene regulation [39][55][69][75][78][79][80][39,55,69,75,78,79,80]?
These questions are relevant to understanding the regulation of the Hox clusters because of the high gene density and compact nature of the clusters. The enhancers embedded within and flanking an individual Hox cluster can display selective preferences, competition between genes and can regulate both near adjacent genes or act more globally on other genes in the complex. For example, in the mouse Hoxb complex, there are three RAREs in the middle of the cluster, two upstream and one downstream of Hoxb4 (Figure 2A), which participate in mediating its response to RA by regulating multiple coding and long non-coding (lncRNAs) transcripts [33][35][37][81][33,35,37,81]. One of these RAREs (DE-RARE) is an essential cis element of an RA-dependent enhancer, which undergoes epigenetic modifications, and is required to coordinate the global regulation of Hoxb genes in hematopoietic stems cells [34]. This functional role for an enhancer raises many questions regarding the mechanisms through which the DE-RARE participates in regulating so many transcripts, how targets are selected and the dynamics of the process. Why, in contrast, do other enhancers embedded in the Hoxb cluster only appear to work on a single near adjacent gene [37][62][82][83][84][37,62,82,83,84]?
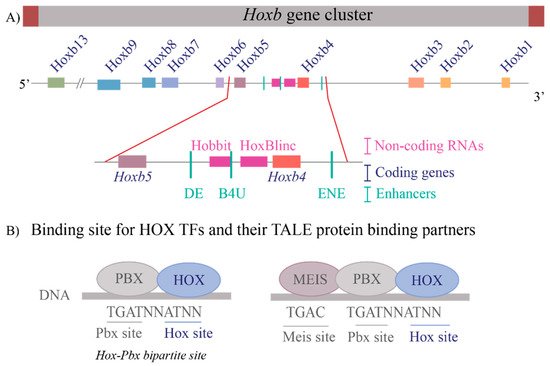
Figure 2. Transcriptional complexity of the Hoxb gene cluster and binding of HOX Transcription factors to DNA (A) A drawing of the Hoxb gene cluster to illustrate that non-coding RNAs as well as enhancers that contain RAREs (Retinoic Acid Response Elements) are interspersed within the coding Hox genes. The enlargement of the Hoxb4-Hoxb5 region shows the complexity within the region that contains three RAREs, two present upstream of Hoxb4 and one present downstream of Hoxb4 and two non-coding RNAs, Hobbit and HoxBlinc. Brown boxes flank the cluster depict boundary elements, colored squares are different Hox genes, pink boxes are non-coding RNAs, and green lines represent RARE enhancers. (B) Depicts the consensus DNA binding sites for HOX proteins and their binding partners, the TALE proteins PBX and MEIS. HOX proteins can bind on Hox-Pbx bipartite sites, or they can bind on DNA in ternary complexes along with both PBX and MEIS. Blue ovals are HOX proteins, and grey ovals are TALE protein binding partners.