Gastric cancer (GC) is the third leading cause of cancer-associated death worldwide. The majority of patients are diagnosed at an advanced/metastatic stage of disease due to a lack of specific symptoms and lack of screening programs, especially in Western countries. Thus, despite the improvement in GC therapeutic opportunities, the survival is disappointing, and the definition of the optimal treatment is still an unmet need. Novel diagnostic techniques were developed in clinical trials in order to characterize the genetic profile of GCs and new potential molecular pathways, such as the Fibroblast Growth Factor Receptor (FGFR) pathway, were identified in order to improve patient’s survival by using target therapies. The aim of this review is to summarize the role and the impact of FGFR signaling in GC and to provide an overview regarding the potential effectiveness of anti-FGFR agents in GC treatment in the context of precision medicine.
- gastric cancer
- FGFR fusions
1. Introduction
2. The FGFR Signaling Pathway and Its Alterations in Gastric Cancer
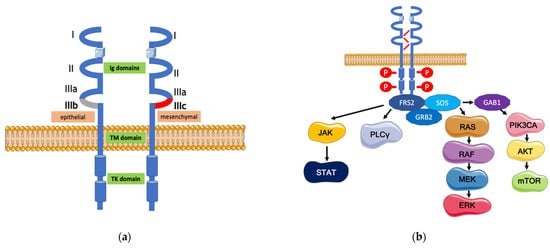
3. A Pharmacological Overview on the Anti-FGFR Agents
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Balakrishnan, M.; George, R.; Sharma, A.; Graham, D.Y. Changing Trends in Stomach Cancer throughout the World. Curr. Gastroenterol. Rep. 2017, 19, 36.
- Luo, G.; Zhang, Y.; Guo, P.; Wang, L.; Huang, Y.; Li, K. Global patterns and trends in stomach cancer incidence: Age, period and birth cohort analysis. Int. J. Cancer 2017, 141, 1333–1344.
- Petrillo, A.; Smyth, E.C. 27 years of stomach cancer: Painting a global picture. Lancet Gastroenterol. Hepatol. 2019, 5, 5–6.
- Tan, P.; Yeoh, K.-G. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 2015, 149, 1153–1162.e3.
- Peek, R.M., Jr.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37.
- Tramacere, I.; Negri, E.; Pelucchi, C.; Bagnardi, V.; Rota, M.; Scotti, L.; Islami, F.; Corrao, G.; La Vecchia, C.; Boffetta, P. A meta-analysis on alcohol drinking and gastric cancer risk. Ann. Oncol. 2011, 23, 28–36.
- Smyth, E.C.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D.; ESMO Guidelines Committee. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27 (Suppl. 5), v38–v49.
- Muro, K.; Van Cutsem, E.; Narita, Y.; Pentheroudakis, G.; Baba, E.; Li, J.; Ryu, M.-H.; Zamaniah, W.I.W.; Yong, W.-P.; Yeh, K.-H.; et al. Pan-Asian adapted ESMO Clinical Practice Guidelines for the management of patients with metastatic gastric cancer: A JSMO–ESMO initiative endorsed by CSCO, KSMO, MOS, SSO and TOS. Ann. Oncol. 2019, 30, 19–33.
- Wagner, A.D.; Syn, N.L.; Moehler, M.; Grothe, W.; Yong, W.P.; Tai, B.C.; Hol, J.; Unverzagt, S. Chemotherapy for advanced gastric cancer. Cochrane Database Syst. Rev. 2017, 8, Cd004064.
- Körfer, J.; Lordick, F.; Hacker, U.T. Molecular Targets for Gastric Cancer Treatment and Future Perspectives from a Clinical and Translational Point of View. Cancers 2021, 13, 5216.
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209.
- Wang, J.; Xiu, J.; Baca, Y.; Battaglin, F.; Arai, H.; Kawanishi, N.; Soni, S.; Zhang, W.; Millstein, J.; Salhia, B.; et al. Large-scale analysis of KMT2 mutations defines a distinctive molecular subset with treatment implication in gastric cancer. Oncogene 2021, 40, 4894–4905.
- Salem, M.E.; Puccini, A.; Xiu, J.; Raghavan, D.; Lenz, H.-J.; Korn, W.M.; Shields, A.F.; Philip, P.A.; Marshall, J.L.; Goldberg, R.M. Comparative Molecular Analyses of Esophageal Squamous Cell Carcinoma, Esophageal Adenocarcinoma, and Gastric Adenocarcinoma. Oncology 2018, 23, 1319–1327.
- Van Cutsem, E.; Bang, Y.J.; Feng-Yi, F.; Xu, J.M.; Lee, K.W.; Jiao, S.C.; Chong, J.L.; López-Sanchez, R.I.; Price, T.; Gladkov, O.; et al. HER2 screening data from ToGA: Targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer 2015, 18, 476–484.
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697.
- Petrillo, A.; Smyth, E.C. Biomarkers for Precision Treatment in Gastric Cancer. Visc. Med. 2020, 36, 364–372.
- Chao, J.; Fuchs, C.S.; Shitara, K.; Tabernero, J.; Muro, K.; Van Cutsem, E.; Bang, Y.-J.; De Vita, F.; Landers, G.; Yen, C.-J.; et al. Assessment of Pembrolizumab Therapy for the Treatment of Microsatellite Instability-High Gastric or Gastroesophageal Junction Cancer Among Patients in the KEYNOTE-059, KEYNOTE-061, and KEYNOTE-062 Clinical Trials. JAMA Oncol. 2021, 7, 895–902.
- Gambardella, V.; Fleitas, T.; Tarazona, N.; Papaccio, F.; Huerta, M.; Roselló, S.; Gimeno-Valiente, F.; Roda, D.; Cervantes, A. Precision Medicine to Treat Advanced Gastroesophageal Adenocarcinoma: A Work in Progress. J. Clin. Med. 2020, 9, 3049.
- Liu, F.-T.; Li, N.-G.; Zhang, Y.-M.; Xie, W.-C.; Yang, S.-P.; Lu, T.; Shi, Z.-H. Recent advance in the development of novel, selective and potent FGFR inhibitors. Eur. J. Med. Chem. 2019, 186, 111884.
- Roskoski, R. The role of fibroblast growth factor receptor (FGFR) protein-tyrosine kinase inhibitors in the treatment of cancers including those of the urinary bladder. Pharmacol. Res. 2019, 151, 104567.
- Roy Burman, D.; Das, S.; Das, C.; Bhattacharya, R. Alternative splicing modulates cancer aggressiveness: Role in EMT/metastasis and chemoresistance. Mol. Biol. Rep. 2021, 48, 897–914.
- Holzmann, K.; Grunt, T.; Heinzle, C.; Sampl, S.; Steinhoff, H.; Reichmann, N.; Kleiter, M.; Hauck, M.; Marian, B. Alternative Splicing of Fibroblast Growth Factor Receptor IgIII Loops in Cancer. J. Nucleic Acids 2011, 2012, 950508.
- Raju, R.; Palapetta, S.M.; Sandhya, V.K.; Sahu, A.; Alipoor, A.; Balakrishnan, L.; Advani, J.; George, B.; Kini, K.R.; Geetha, N.P.; et al. A Network Map of FGF-1/FGFR Signaling System. J. Signal Transduct. 2014, 2014, 962962.
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569.
- Del Piccolo, N.; Sarabipour, S.; Hristova, K. A New Method to Study Heterodimerization of Membrane Proteins and Its Application to Fibroblast Growth Factor Receptors. J. Biol. Chem. 2017, 292, 1288–1301.
- Ferguson, H.R.; Smith, M.P.; Francavilla, C. Fibroblast Growth Factor Receptors (FGFRs) and Noncanonical Partners in Cancer Signaling. Cells 2021, 10, 1201.
- Ong, S.H.; Guy, G.R.; Hadari, Y.R.; Laks, S.; Gotoh, N.; Schlessinger, J.; Lax, I. FRS2 Proteins Recruit Intracellular Signaling Pathways by Binding to Diverse Targets on Fibroblast Growth Factor and Nerve Growth Factor Receptors. Mol. Cell. Biol. 2000, 20, 979–989.
- Arkun, Y.; Yasemi, M. Dynamics and control of the ERK signaling pathway: Sensitivity, bistability, and oscillations. PLoS ONE 2018, 13, e0195513.
- Mossahebi-Mohammadi, M.; Quan, M.; Zhang, J.-S.; Li, X. FGF Signaling Pathway: A Key Regulator of Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 79.
- Mandal, S.; Bandyopadhyay, S.; Tyagi, K.; Roy, A. Recent advances in understanding the molecular role of phosphoinositide-specific phospholipase C gamma 1 as an emerging onco-driver and novel therapeutic target in human carcinogenesis. Biochim. Biophys. Acta Rev. Cancer. 2021, 1876, 188619.
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496.
- Gu, W.; Yang, J.; Wang, Y.; Xu, J.; Wang, X.; Du, F.; Hu, X.; Guo, H.; Song, C.; Tao, R.; et al. Comprehensive identification of FGFR1-4 alterations in 5 557 Chinese patients with solid tumors by next-generation sequencing. Am. J. Cancer Res. 2021, 11, 3893–3906.
- Sun, Y.; Li, G.; Zhu, W.; He, Q.; Liu, Y.; Chen, X.; Liu, J.; Lin, J.; Han-Zhang, H.; Yang, Z.; et al. A comprehensive pan-cancer study of fibroblast growth factor receptor aberrations in Chinese cancer patients. Ann. Transl. Med. 2020, 8, 1290.
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456.
- Kuboki, Y.; Schatz, C.A.; Koechert, K.; Schubert, S.; Feng, J.; Wittemer-Rump, S.; Ziegelbauer, K.; Krahn, T.; Nagatsuma, A.K.; Ochiai, A. In situ analysis of FGFR2 mRNA and comparison with FGFR2 gene copy number by dual-color in situ hybridization in a large cohort of gastric cancer patients. Gastric Cancer 2017, 21, 401–412.
- Matsumoto, K.; Arao, T.; Hamaguchi, T.; Shimada, Y.; Kato, K.; Oda, I.; Taniguchi, H.; Koizumi, F.; Yanagihara, K.; Sasaki, H.; et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br. J. Cancer 2012, 106, 727–732.
- Lee, S.J.; Hong, J.Y.; Kim, K.; Kim, K.-M.; Kang, S.Y.; Lee, T.; Kim, S.T.; Park, S.H.; Park, Y.S.; Lim, H.Y.; et al. Detection of Fusion Genes Using a Targeted RNA Sequencing Panel in Gastrointestinal and Rare Cancers. J. Oncol. 2020, 2020, 4659062.
- Costa, R.; Carneiro, B.A.; Taxter, T.; Tavora, F.A.; Kalyan, A.; Pai, S.A.; Chae, Y.K.; Giles, F.J. FGFR3-TACC3 fusion in solid tumors: Mini review. Oncotarget 2016, 7, 55924–55938.
- Kim, S.Y.; Ahn, T.; Bang, H.; Ham, J.S.; Kim, J.; Kim, S.T.; Jang, J.; Shim, M.; Kang, S.Y.; Park, S.H.; et al. Acquired resistance to LY2874455 in FGFR2-amplified gastric cancer through an emergence of novel FGFR2-ACSL5 fusion. Oncotarget 2017, 8, 15014–15022.
- Zhou, Y.; Wu, C.; Lu, G.; Hu, Z.; Chen, Q.; Du, X. FGF/FGFR signaling pathway involved resistance in various cancer types. J. Cancer 2020, 11, 2000–2007.
- Nienhüser, H.; Schmidt, T. Angiogenesis and Anti-Angiogenic Therapy in Gastric Cancer. Int. J. Mol. Sci. 2017, 19, 43.
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.-C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694.
- Facchinetti, F.; Hollebecque, A.; Bahleda, R.; Loriot, Y.; Olaussen, K.A.; Massard, C.; Friboulet, L. Facts and New Hopes on Selective FGFR Inhibitors in Solid Tumors. Clin. Cancer Res. 2019, 26, 764–774.
- de Almeida Carvalho, L.M.; de Oliveira Sapori Avelar, S.; Haslam, A.; Gill, J.; Prasad, V. Estimation of Percentage of Patients with Fibroblast Growth Factor Receptor Alterations Eligible for Off-label Use of Erdafitinib. JAMA Netw. Open. 2019, 2, e1916091.
- Weaver, A.; Bossaer, J.B. Fibroblast growth factor receptor (FGFR) inhibitors: A review of a novel therapeutic class. J. Oncol. Pharm. Pract. 2020, 27, 702–710.
- Perera, T.P.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and Pharmacological Characterization of JNJ-42756493 (Erdafitinib), a Functionally Selective Small-Molecule FGFR Family Inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020.
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348.
- Liu, P.C.C.; Koblish, H.; Wu, L.; Bowman, K.; Diamond, S.; DiMatteo, D.; Zhang, Y.; Hansbury, M.; Rupar, M.; Wen, X.; et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS ONE 2020, 15, e0231877.
- Vogel, A.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. LBA40—FIGHT-202: A phase II study of pemigatinib in patients (pts) with previously treated locally advanced or metastatic cholangiocarcinoma (CCA). Ann. Oncol. 2019, 30, v876.
- Xiang, H.; Chan, A.G.; Ahene, A.; Bellovin, D.I.; Deng, R.; Hsu, A.W.; Jeffry, U.; Palencia, S.; Powers, J.; Zanghi, J.; et al. Preclinical characterization of bemarituzumab, an anti-FGFR2b antibody for the treatment of cancer. mAbs 2021, 13, 1981202.