Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yuliya Kulikova and Version 2 by Vicky Zhou.
At the moment, macroalgae blooms in sea waters, the rotting of which causes greenhouse gas emissions and contributes to the formation of a negative ecological and economic situation in coastal zones, which has become a serious problem. Fuel production through hydrothermal liquefaction (HTL) of macroalgae and marine debris is a promising solution to this ecological problem.
- hydrothermal liquefaction
- macroalgae
- bio-oil
- biomass
- plastic
- polymers
- marine waste
1. Introduction
Nowadays bursts of macroalgae development have become a frequent occurrence and are associated with an increase in anthropogenic load on the hydrosphere objects and climate change. Objectively, there has been a three-fold increase in the number of recorded cases over the past 30 years [1]. Rotting algae becomes a source of greenhouse gases, creating a secondary anthropogenic load on the biosphere. The potential for the formation of greenhouse gases for algae is relatively high and amounts to 498.75 mL per 1 g of dry matter [2].
At the same time, the biomass of macroalgae has significant resource potential [3][4][3,4]. There are some articles on macroalgae processing with the production of bioethanol [5], biogas [6][7][6,7], biofuels [2][8][9][2,8,9] nutrients [10], and biologically active substances [11].
It should be noted that the processing of drifting macroalgae into fuel will allow for solving two problems of reducing the climatic load at once: namely, it will ensure the prevention of methane formation in the decay process and will make it possible to advance in solving the key problem of modern society—the reduction of CO2 emissions from the use of fossil fuels [12] and their replacement with carbon-neutral sources.
This article is focused on recycling drifting algae that washed ashore and has become, in fact, waste. Such algae, together with marine garbage, are collected from the beaches by special sieving machines and stored in heaps until they are taken to a landfill. The accumulation of such biomass varies significantly depending on the season and geography of the coast.
The problem of processing algae that has both drifted and washed ashore in order to produce fuel is due to their high moisture content, composition variability, seasonal fluctuations in volumes, and high degree of contamination with foreign impurities.
Technologies for direct conversion of biomass into liquid fuel can be conditionally divided into two groups: biochemical and thermochemical. The most suitable processing method depends on the amount and type of biomass, the desired form of energy produced, end-use requirements, environmental standards, and economic feasibility [13][14][13,14].
The major problems associated with the biochemical production of biofuels from algae are low fuel yield (no more than 50%) [15][16][17][15,16,17], the need for preliminary preparation of biomass, and the high cost of extracting target products [18][19][18,19].
Direct thermochemical conversion of algae biomass into liquid fuel involves pyrolysis and hydrothermal liquefaction. The main problem of producing biofuel from biomass by pyrolysis is associated with the need to dry raw materials to a moisture content of no more than 10%, which significantly worsens the technical, economic, and energy parameters of the process [20]. Due to the heterogeneity of raw materials, there is a risk of poisoning catalytic systems [21], and the fuel yield does not exceed 30%.
Hydrothermal liquefaction (HTL) refers to a thermochemical process in which biomass and organic waste are decomposed by the action of supercritical (i.e., the water pressure and temperature are above the critical point of 374 °C and 22.1 MPa) or subcritical water (i.e., when the water temperature is significantly below the critical point). Under such conditions, water possesses certain unique properties. For example, near the critical point, the dissociation constant of water increases sharply, forcing water to act as an acidic or basic catalyst for numerous reactions which makes it possible to chemically transform almost all types of organic compounds through hydrolysis, ionic, or radical reactions [12]. Reactions proceed in sub- and supercritical water in fundamentally different ways. In general, ionic reactions are peculiar to the hydrothermal liquefaction in the subcritical water conditions, whereas reactions occurring in supercritical water are the free-radical reactions [12]. Despite said general dependency of the reaction type on the water conditions, there are studies reporting that ionic reactions can still happen in supercritical water, as was shown by Ciuffi et al. [22].
The process is typically carried out in an aqueous medium at temperatures ranging from 250 °C to 380 °C and pressures ranging from 4 to 25 MPa [23]. The usual dry matter content in the introduced mixture is from 5% to 20% [24], so the biomass can be processed without preliminary drying.
Hydrothermal liquefaction, like conventional pyrolysis, yields three main products: bio-oil, solid residue, and gas phase, as well as an aqueous phase containing soluble products. Liquid organic fraction (bio-oil) is obviously the most valuable product among them. In this regard, the majority of current research is devoted to the search for methods to increase the yield of bio-oil, including the use of catalysts [25][26][25,26], the selection of solvents for the extraction of bio-oil from an aqueous solution [27], and the joint processing of various types of raw materials [28].
Using the HTL process typically results in higher quality fuel after drying. As a result, the fuel has a lower oxygen content and a higher calorific value (HHV 33–36 MJ/kg) and thus requires much less effort to condition than pyrolysis oil [29]. According to the literature, hydrothermal liquefaction [30][31][32][33][30,31,32,33] can also provide fuel with a lower cost price of $0.57–1.2 per L (for gasification processes this figure is 1.1–1.73 [12] and for pyrolysis is 1.54–1.78 [12]), while the fuel will be of reasonably good quality. The ability to process wet raw materials with a moisture content of up to 80% (for example, sewage sludge, manure, algae, etc.) without the need for drying or other pretreatment is one of the benefits of HTL [34]. Thus, the HTL process should be considered the most promising for the direct transformation of wet algal biomass.
According to the literature data, a mixture of drifting macroalgae is inevitably contaminated with impurities of foreign fractions to one degree or another. The world’s total plastic reserves in the oceans amount to five trillion elements weighing more than 0.25 million tons, while in the surface layer of water, the concentration of plastic reaches half a million elements per square kilometer [35]. Plastic marine debris significantly affects the quality and safety of the aquatic environment, posing a threat to aquatic organisms and human health, and becoming a problem on the path to ensuring food safety [32][33][34][35][36][37][38][39][32,33,34,35,36,37,38,39].
Polymers account for approximately 60–90% of the total mass of marine waste, according to an analysis of the component composition of marine debris [38][39][38,39]. Typical polymers and polymer-containing composite in marine debris are high- and low-pressure polyethylene, polyethylene terephthalate, Tetra Pak, polypropylene, and nylon [39][40][39,40].
There are few studies on the hydrothermal liquefaction of polymers or their mixtures [40][41][42][43][44][45][46][47][48][49][40,41,42,43,44,45,46,47,48,49], and only a few studies are devoted to the joint recovery of biomass of macroalgae and polymers to produce liquid fuel [50][51][50,51].
2. Prospects for Joint Processing of Macroalgae Biomass and Marine Debris
There are few studies on the HTL of polymers or their mixtures [40][41][42][43][44][45][46][47][48][49][40,41,42,43,44,45,46,47,48,49] and only two studies are devoted to the joint recovery of macroalgae and polymers with the production of liquid fuel [50][51][50,51]. In this connection, it was decided to consider the results of studies pertaining to the HTL of polymers with/or without adding other types of biomasses and to assess the perspectives of their joint processing with algae. Table 13 presents an overview of current research in the field of HTL of polymers and their mixtures with biomass.Table 13.
Overview of polymer processing experiments.
| Polymers | Conditions | Fuel Yield Efficiency | HHV of the Oil | References | |||||
|---|---|---|---|---|---|---|---|---|---|
| High- and low-pressure polyethylene | Temperature 400–450 °C, 0.5 and 4 h. Raw material dose 1:1.75 | The maximum yield (97 wt%) of a mixture of paraffinic and α-olefin waxes was obtained at 425 °C and 30–40 min. The maximum fuel yield (87 wt%) was obtained at 425 °C, 2.5 h or 450 °C, 45 min. | 42.7–42.9 MJ/kg | [43] | |||||
| Polypropylene and polyethylene | Pressure 0.25, 1.55, 3.75, 10.25, and 23 MPa. Polymer: water ratio 1:1.75; 200:1; 7.3:1; 2.3:1, time 45–60 min, temperature 450 °C | The minimum coal yield and the optimum degree of destruction were achieved at a water content of 5% and 1.55 MPa. The light ends of the fuel, obtained from a mixture of PP:PE in a 1:1 ratio, meet all the requirements for high-quality pure gasoline, and the heavy ends meet the requirements for ultra-low sulfur diesel fuel. | 42.6–42.7 MJ/kg for oil from polyethylene and 42.2–42.4 MJ/kg for oil from polypropylene | [44] | |||||
| Tetra Pak | Temperature 300–420 °C, pressure 16–24 MPa, time 5–60 min, and polymer concentration 5–40%. | The maximum fuel yield of 35.55% was achieved at 360 °C, 22 MPa, 30 min and a feed concentration of 20 wt%. The maximum fuel calorific value of 48.747 MJ/kg and the maximum energy extraction efficiency of 46.49% were observed at 420 °C, 20 MPa, 30 min and a feed concentration of 20 wt% | The maximum fuel HHV 48.747 MJ/kg and the maximum energy extraction efficiency of 46.49% were observed at 420 °C, 20 MPa, 30 min and a feed concentration of 20 wt%. | [45] | |||||
| 12 polymers: polyacrylonit-rylbutadiene-styrene (ABS), bisphenol-A epoxy resin, high pressure polyethylene, low pressure polyethylene, polyamide 6 polyamide 66, polyethylene terephthalate, polycarbonate, polypropylene, polystyrene and polyurethane foam, polyvinyl chloride. | Temperature 350 °C, exposure time 14 min and 1:17 (polymer:water ratio), catalyst KOH 17.2 g/L | LDPE, HDPE, PP, and PS showed a solid yield of more than 90%, the formation of oil did not exceed 5%. The catalyst allowed increasing the destruction of ABS, epoxy resin, polyamides, PET, polycarbonate, and polyurethane foam to achieve an average solid fraction yield of 0–25%, while without a catalyst this value was 50–86%. | n/a | [46] | |||||
| Polybutylene terephthalate, polycarbonate, polyethylene terephthalate, polylactate, polymethyl methacrylate, polyoxymethylene, styrene butadiene (SBD), polyvinyl acetate (PVA) | Temperature 400 °C, 25 MPa, 15 min, raw material dose 1:10 | The synthetic crude oil yield ranged from 0% (for PET, PBT, polylactate) and almost 100% for PC. SBD and PVA also demonstrated high biofuel yield up to 80% | n/a | [42] | |||||
| Polypropylene, polystyrene, polycarbonate, and polyethylene terephthalate. | Time: 0.5 h, 1 h, temperature 350–450 °C, pressure 16.52 and 25 MPa | The maximum yield was from 16 wt% for PET and up to 86 wt% for polystyrene. Depolymerization of plastics to oil was fastest under supercritical conditions (T > 400 °C). | 30–46 MJ/kg for oil from polypropylene; 36–46 MJ/kg for oil from polystyrene; 28–30 MJ/kg for oil from polycarbonate; 28–29 MJ/kg for oil from PET | [47] | |||||
| Polymer mixture: cellulose, PET, nylon, PVA, polyethylene, polypropylene | Temperature 340 °C, 5 h, raw material dose 1:10, catalyst 2% NaOH (for PET hydrolysis) | Comparison of the results of the two tests showed that the addition of NaOH led to a decrease in the percentage of solid residue (from 75.1% to 65.5%) and an increase in the percentage of water-soluble organic substances (from 6.2% to 16%), the fuel yield remained practically the same (7.7% and 7.4%, respectively). | n/a | [48] | |||||
| A mixture of pistachio shells, polyethylene, PET, polypropylene, nylon-6 | The weight ratios of biomass and plastic are 100:0, 90:10, and 80:20. Temperature 350 °C, time 15, and 60 min. |
Polyolefins led to a decrease in bio-oil yield from 34% to 20%, PET and nylon provided a slight increase in oil yield to 36.1 and 34.6%, respectively. | 34 MJ/kg for oil from pistachio shells; shells with 10–20% of PET 33–35 MJ/kg; shells with 10–20% of PE 37–38 MJ/kg; shells with 10–20% of PP 32–33 MJ/kg; shells with 10–20% of nylon 32.5–33 MJ/kg. | [52] | [88] | ||||
| Biomass of | Prosopis juliflora | shrub and polyolefins | Biomass: Polymers Ratio: 0:1, 1:0, 1:1, 2:1, 3:1, 4:1, and 5:1, temperature 340–440 °C, catalyst: bentonite clay activated with hydrochloric acid | The maximum bio-oil yield was about 61.23 wt% at 420 °C for a 3:1 mixture with the addition of 3 wt% catalyst with a holding time of 60 min. | 45.2 MJ/kg for oil from | Prosopis juliflora | ; 44.18 MJ/kg for oil from polyolefins; 46.0 MJ/kg for oil from blend 3:1 | [53] | [89] |
| Cellulose, starch, soy protein isolate, lignin, stearic acid, glucose and various polymers: polypropylene, polystyrene, polycarbonate, and polyethylene terephthalate | Time: 0.5 h, temperature 300, 350, 400, 425 °C, pressure 25 MPa | Increase in oil yield by two (45%), with joint processing with polystyrene, decrease in the decomposition temperature of polyolefins (up to 300 °C). | 27.9–32.1 MJ/kg for oil from biomolecules blend; 36.4–38.5 MJ/kg for oil from polymers blend; 30.9–34.0 MJ/kg for oil from 9 components blend (polymers and biomolecules) | [54] | [90] | ||||
| A mixture of macroalgae and plastic marine debris (polyethylene, polypropylene, nylon) | Temperature 340 °C, pressure 16.5 MPa, time 12 and 27 min, plastic dose 0, 10%, 25%, 50% of the mixture mass | The biofuel yield did not exceed 15%, but with the addition of plastics, a significant increase was noted (from 10% to 15%), the calorific value of the fuel increased from 33.5 to 39.0 MJ/kg | With the addition of plastics, a significant increase was noted (from 10% to 15%), HHV of the fuel increased from 33.5 to 39.0 MJ/kg | [55] | [91] | ||||
| A mixture of | HHV of oil from algae 33 MJ/kg, HHV of oil from algae+ nylon 28–35 MJ/kg | [ | 49 | ] |
2.1. Hydrothermal Liquefaction of Polymers and Their Mixtures
Pedersen and Conti [42] assessed the prospects of using the HTL process for processing nine types of solid polymers, including polybutylene terephthalate, polycarbonate, polyethylene terephthalate, polylactate, polymethyl methacrylate, polyoxymethylene, styrene butadiene, polyvinyl acetate, and polyphenylene oxide.
The process was carried at the temperature of 400 °C and pressure of 25 MPa for 15 min and the mass dose of raw materials was 10%. The polymers have shown that they are readily degraded in the HTL process, with the formation of water-soluble organic compounds, biofuels, gaseous products, and a solid residue. Figure 12 shows the average oil and solid matter yield for each polymer [42]. Analysis of the data establishes that the processing of a typical representative of marine debris, polyethylene terephthalate, even under supercritical conditions (400 °C, 25 MPa) did not lead to the formation of a liquid fraction, almost the entire volume of carbon passed into a solid phase (biochar). Good fuel yield was obtained during processing of polycarbonate, styrene butadiene, and polyphenylene oxide, but the proportion of these polymers in marine debris is negligible.
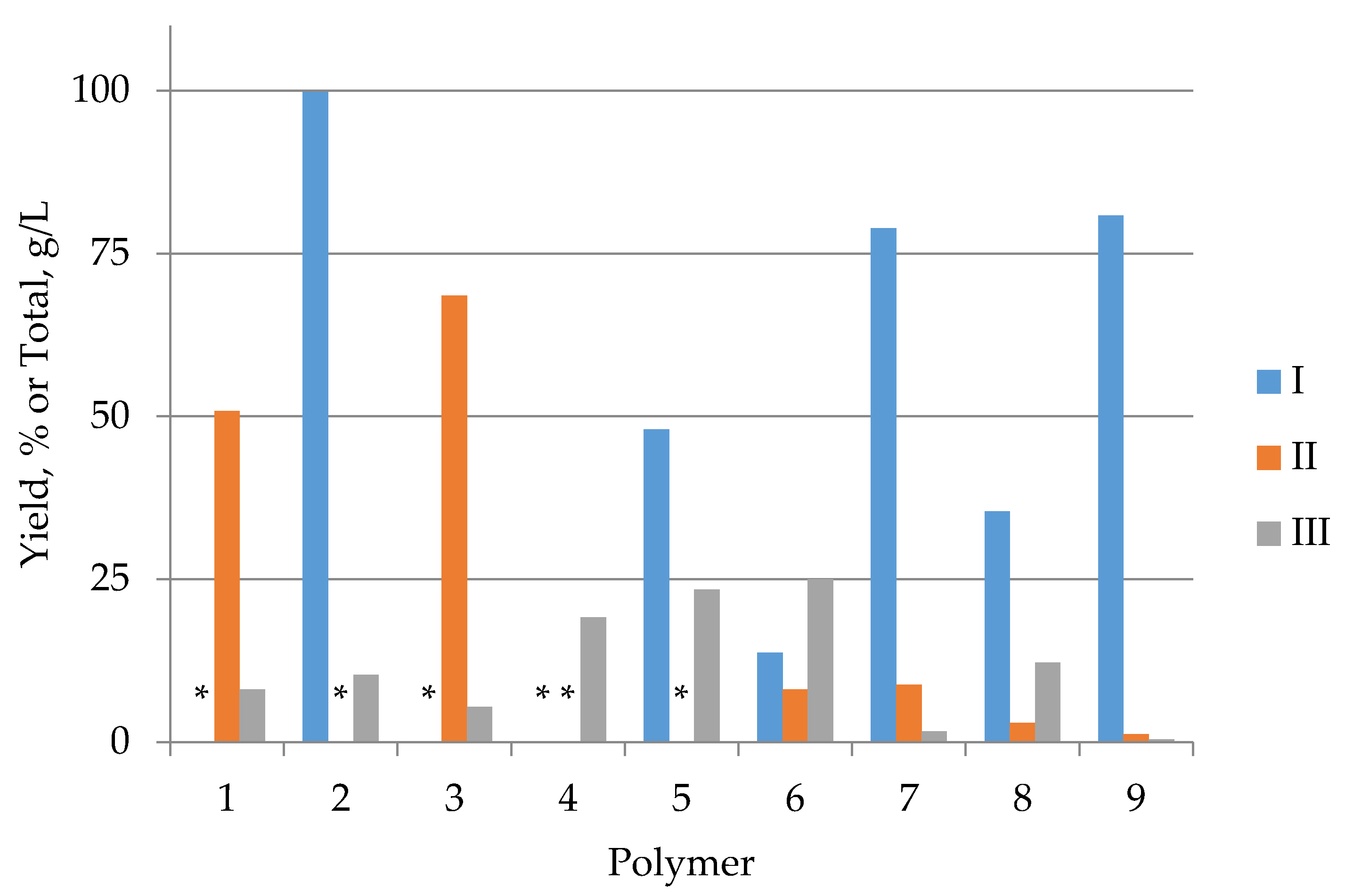
Figure 12. The results of polymer processing by the hydrothermal liquefaction: I—oil yield, %; II—Solid Phase Yield, %; III—Total Organic Carbon in the Aqueous Phase, g/L. Plymers: 1—Polybutylene terephthalate; 2—Polycarbonate; 3—PET; 4—Polylactate; 5—Polymethyl methacrylate; 6—Polyoxymethylene; 7—Polyphenylene oxide; 8—Polyvinyl acetate; 9—Styrene butadiene. *—no data.
Jin et al. [43] studied in detail the effect of temperature and reaction time on the degradation of polyethylene. As a result of the study, it was found that during the process (up to 4 h) at temperatures below 400 °C, the formation of liquid biofuel did not occur. At a temperature of 425 °C, the total yield of fuel and paraffins varied from 69% to 87%, reaching a maximum at a process time of 2.5 h. With time increase the yield of the gas phase increased from 1% to 21% at process times of 1 and 4 h, respectively. At 450 °C, a similar maximum fuel yield was achieved with a process time of 45 min.
Another research group obtained the maximum yield (97 wt%) of paraffinic and α-olefin waxes mixture during the HTL processing of polyethylene at 425 °C and a reaction time of 30–40 min, while the solid fraction all experiments was not found [42].
Jin et al. processed polypropylene at a temperature of 425 °C and achieved a fuel yield of 91 wt% after 2 h, which indicates a faster conversion of PP than from PE. The polypropylene fuel contained more olefins and cycloparaffins (about 90 wt%) and less paraffins (about 10 wt%) [44].
A research group from Xi’an University of Technology processed Tetrapak at different temperatures and found that the bio-oil yield increased from 4.56% to 24.34% when the temperature was raised from 300 °C to 360 °C. However, temperatures above 360 °C had some negative impacts on bio-oil yield. This is mainly due to the following: firstly, the higher temperature promoted the gasification reaction; secondly, at higher temperatures, polyethylene will envelop the paper, reducing porosity and preventing heat and mass transfer [45].
A research group from Aarhus University evaluated the prospects of using the HTL process for processing a wide range of polymers [46]. The process was carried out at 350 °C for 14 min in the presence of a KOH catalyst of 17.2 g/L. The main findings of the study regarding polymers typical of marine debris can be summarized as follows:
-
PET behaved very different from others, since upon addition of a catalyst the formed insoluble terephthalic acid transformed into a soluble salt, increasing the COD of the aqueous phase [46].
-
PVC was the only polymer that exhibited carbonization reactions rather than liquefaction reactions. The addition of alkali did not lead to significant differences in the yields of oil and char; however, a large difference was noted in the volumes of the gas phase. Treatment of PVC in the presence of alkali gives an increased gas yield due to the release of Cl2 amount. Solid residues of PVC were strongly dechlorinated, which indicates that this fraction can be further used as a carbon source [46].
| Fucus serratus |
| macroalgae and nylon |
| 350 °C during 10 min, 4 grades of nylon: nylon 6, nylon 6/6, nylon 12, and nylon 6/12 in a mixture of 5, 20, and 50 wt% nylon in relation to biomass |
| 100% of nylon 6 and 6/6 degrades under HTL conditions while forming cyclopentanone. |
| Nylon 6/12 and nylon 12 were less reactive, only traces of monomer were observed |
- In general, each type of synthetic polymer exhibits its own depolymerization characteristics at HTL, which creates opportunities and challenges for future applications for net and mixed streams. Short exposure time of subcritical HTL was not efficient for polyolefins and polystyrene, however it could be useful for polymers, which contain heteroatoms.
A group of scientists from Pennsylvania State University assessed the effect of the processing time (0.5 h, 1 h) and temperature (350–450 °C) on the yield, composition, and quality of oil obtained as a result of HTL of polypropylene, polystyrene, polycarbonate and polyethylene terephthalate. The maximum oil yield for each material varied from 16 wt% for PET to 86 wt% for polystyrene. Depolymerization of plastics occurred fastest in supercritical water (T > 400 °C). Longer reaction times and higher supercritical temperatures reduce the fuel yield but increase its calorific value (HHV) [47].
2.2. Joint Hydrothermal Liquefaction of Polymers and Biomass
Joint processing of pistachio shells and a mixture of the four most common polymers (PET, polyethylene, polypropylene, and nylon) caused an increase of solid yields compared to HTL of pure pistachio shells. Co-processing of polyethylene and polypropylene decreased oil yield from 34.1% to 20.4% and from 34.1% to 30.7%. The yield of bio-oil during processing with PET remained practically unchanged, while the yield of char increased by a multiple of the number of added polymers [52][88].
Unlike other plastics, the increase in biochar yield was much less when pistachio shells were co-liquefied with nylon (the maximum solids increase was 9% with the addition of 20% polymer). The biofuel yield for nylon mixes was almost the same and amounted to 30.0%, while a significant change in the composition of the aqueous phase was observed. This behavior of nylon has been confirmed by other studies [52][88] and is associated with the transformation of nylon during HTL processing into water-soluble caprolactam.
The co-processing of pistachio shells with a mixture of plastics in equal proportions led to a significant increase in bio-char production, while the bio-oil yield decreased by 20%. This fact, according to the authors, confirmed the lack of expediency with the joint processing of biomass and polymers regarding bio-oil production.
Savage et al. [54][90] studied the synergistic effect of the combined processing of cellulose, starch, soy protein isolate, lignin, stearic acid, glucose, and various polymers, including polypropylene, polystyrene, polycarbonate, and polyethylene terephthalate. They found that the combined processing of cellulose, starch, and lignin with plastics provided an increase in the fuel yield by 13.9 wt% (at 300 °C, 30 min). It was also established that polyolefins were activated during the joint processing of these components, which ensured a decrease in the temperature of their decomposition.
When processing the biomass of shrubs and a mixture of polyolefin waste, it was found that there was a synergistic effect that provided an increase in the average fuel yield from 40% to 50%. The addition of 3 wt% catalyst (bentonite clay activated with hydrochloric acid) provided the maximum bio-oil yield—61.23 wt% (temperature 420 °C for a mixture of three parts of biomass: one part polymer, holding time 60 min) [53][89].
There is little research on the co-processing of macroalgae with polymers typical of marine debris. Detailed studies assessing the effect of several of the most common polymer contaminants (polyethylene, PET, polypropylene, nylon) on the hydrothermal liquefaction (HTL) process of four species of macroalgae (L. digitata, U. lactuca, F. serratus, and S. muticum) were carried out by Raikova et al. [51].
The biomass to polymer ratios were 100:0, 90:10, 75:25, 50:50, and 0:100. The process was carried out at a temperature of (340 ± 3) °C, the reaction time was 12–27 min, and the pressure inside the reactor was 15 MPa. It was found that polyethylene and propylene practically did not decompose under the influence of subcritical water. The presence of all types of plastics in the processed biomass resulted in a moderate synergistic effect which led to an increase in the amount of carbon in biofuels, a decrease in nitrogen content, and an increase in heating value. Nylon (fishing nets) was depolymerized almost completely under the HTL process to form caprolactam, which entered mainly into the aqueous phase. Disadvantageous for biofuel production, the disposal of marine nylon debris to produce monomers still could be a promising source of income for future biofuel plants [49].
To summarize, it can be seen from the results of joint processing of polymers and various types of biomasses that polymers transformed at higher temperatures than biomass [47][53][54][47,89,90]. The addition of polymers does not increase, but rather decreases the yield of biofuels and increases the yield of char, which is undesirable. Thus, the presence of polymers in the biomass of drifting macroalgae should be regarded as a ballast fraction, which, in the case of significant content, can have a negative effect on the process.