Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Masaru Tanaka and Version 3 by Lindsay Dong.
Migraine is a primary headache disorder characterized by a unilateral, throbbing, pulsing headache, which lasts for hours to days, and the pain can interfere with daily activities. It exhibits various symptoms, such as nausea, vomiting, sensitivity to light, sound, and odors, and physical activity consistently contributes to worsening pain.
- primary headache
- migraine
- trigeminal system
- neuropeptides
- neurogenic inflammation
- animal model
- inflammatory soup
- dura mater
- immune system
- migraine treatment
1. Neurogenic Inflammation
The localized form of inflammation is neuroinflammation, which occurs in both the peripheral and CNSs. The main features of NI are the increased vascular permeability, leukocyte infiltration, glial cell activation, and increased production of inflammatory mediators, such as cytokines and chemokines [1][85]. NI increases the permeability of the blood to the brain barrier, thus allowing an increased influx of peripheral immune cells into the CNS [2][86] (Figure 12A).
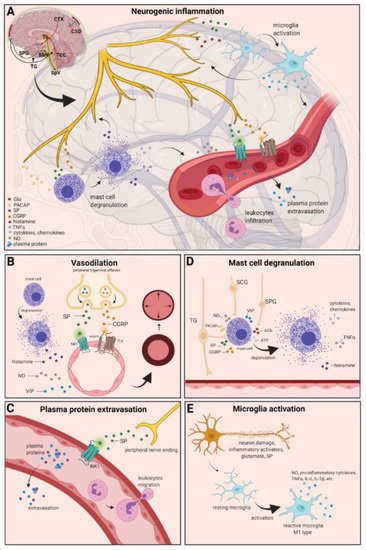
Figure 12. Neurogenic inflammation and its main features. (A) Stimulation of the trigeminal nerve causes the release of neuropeptides, including CGRP, SP, NO, VIP, and 5-HT, leading to neurogenic inflammation, which has four main features: the increased vascular permeability, leukocyte infiltration, glial cell activation, and increased production of inflammatory mediators, such as cytokines and chemokines. (B) Vasoactive peptides, such as CGRP and SP, bind their receptors on smooth muscle of dural vessels and cause vasodilation. The released neuropeptides induce mast cell degranulation, resulting in the release of histamine, which leads endothelium-dependent vasodilation. (C) Binding of the released SP to the NK1 receptors expressed on the microvascular blood vessels disrupts the membrane and causes plasma protein leakage and leukocyte extravasation. (D) Mast cells are in close association with neurons, especially in the dura, where they can be activated following trigeminal nerve and cervical or sphenopalatine ganglion stimulation. Release of neuropeptides causes mast cell degranulation, which leads to release of histamine and serotonin and selectively can cause the release of pro-inflammatory cytokines, such as TNF-α, IL-1, and IL-6. (E) Under the influence of inflammatory stimuli, microglia can become reactive microglia. Microglia activation leads to the production of inflammatory mediators and cytotoxic mediators.
The concept of NI was introduced by the experiment of Goltz and Bayliss, in which skin vasodilation was observed during electrical stimulation of the dorsal horn, which could not be linked to the immune system [3][4][87,88]. Dalessio was the first who hypothesized a connection between NI and migraine and believed that a headache is a result of vasodilation of cranial vessels associated with local inflammation [5][89]. This theory was later reworked by Moskowitz, who believed that upon activation, the neuropeptide release from trigeminal neurons has a role in increasing vascular permeability and vasodilation [6][90].
There are several theories concerning the mechanism of NI. Hormonal fluctuations or cortical spreading depression can initiate two types of processes: activating the TS to trigger the liberation of neuropeptides from the peripheral trigeminal afferents and/or degranulating the mast cells that can lead to the release of neuropeptides by activating and sensitizing the nociceptors [7][91]. In rats, Bolay and colleagues demonstrated that after local electrical stimulation of the cerebral cortex, CSD is generated, and it can trigger trigeminal activation, which causes meningeal inflammation occurring after the CSD disappearance [8][92]. Both CGRP and SP play an important role in the development of NI. Released peptides, such as CGRP, bind to its receptor on smooth muscle cells, eliciting a vasodilatory response, thereby increasing meningeal blood flow in the dural vasculature. In contrast to CGRP, binding of the released SP to the NK1 receptors expressed on the microvascular blood vessels disrupts the membrane and causes plasma protein leakage. Both neuropeptides can induce mast cell degranulation through their specific receptors and further sensitize meningeal nociceptors [7][91]. The meningeal nerve fibers also contain neurotransmitters (e.g., glutamate, serotonin) and hormones (e.g., prostaglandins) that can affect the activation and release of neuropeptides, causing neurogenic inflammation [5][89]. Moreover, several cell types (e.g., endothelial cells, mast cells, and dendritic cells) can release tumor necrosis factor alpha (TNFα), interleukins, nerve-growth factor (NGF), and VIP, also causing plasma protein extravasation (PPE) [9][10][93,94], which is a key characteristic of NI. In addition, neuronal nitric oxide synthase (nNOS) enzyme can be detected in the trigeminal nerve endings, the dural mastocytes, and also the TNC and the TG [11][95], which catalyzes the synthesis of retrograde signaling molecule nitric oxide (NO). NO has a major role in mediating many aspects of inflammatory responses; NO can affect the release of various inflammatory mediators from cells participating in inflammatory responses (e.g., leukocytes, macrophages, mast cells, endothelial cells, and platelets) [12][96]. Through its retrograde signaling action, astrocytes can influence the release of CGRP, SP, and glutamate [13][14][97,98]. Beside this, bradykinin and histamine induce NO release from vascular endothelial cells, suggesting a strong interaction between NO and inflammation [15][99]. The inflammation can lead to CGRP release from the activated primary afferent neurons, which force satellite glial cells to release NO. NO can induce nNOS, which can be considered a significant marker of the sensitization process of the TS. TRPV channels permit afferent nerves to detect thermal, mechanical, and chemical stimuli, thereby regulating NI and nociception [16][100]. TRPV1 was identified in dorsal root ganglion (DRG), TG neurons, and spinal and peripheral nerve terminals [17][101]. Inflammatory mediators remarkably up-regulate TRPV1 through activation of phospholipase C (PLC) and protein kinase A (PKA) and protein kinase C (PKC) signaling pathways [18][19][20][21][102,103,104,105]. Increased TRPV1 expression in peripheral nociceptors contributes to maintaining inflammatory hyperalgesia [17][101]. In an experimental injury model, Vergnolle et al. demonstrated that a decrease in osmolarity of extracellular fluid could induce neurogenic inflammation, which TRPV4 can mediate [22][106]. Furthermore, plasma and cerebrospinal fluid levels of neuropeptides, histamine, proteases, and pro-inflammatory cytokines (e.g., TNFα, IL-1β) are elevated during migraine attacks [23][24][107,108], suggesting that neuroimmune interactions contribute to migraine pathogenesis.
1.1. Vasodilation
There are various cell types in blood vessels that both release and respond to numerous mediators that can contribute to migraine; this includes growth factors, cytokines, adenosine triphosphate (ATP), and NO [25][26][27][28][109,110,111,112]. In the central system, NO may be involved in the regulation of cerebral blood flow and neurotransmission [29][59]. NO can stimulate the release of neuropeptides and causes neurogenic vasodilation [30][113]. In addition to NO, NGF also increases the expression of CGRP and enhances the production and release of neuropeptides, including SP and CGRP, in sensory neurons [31][114]. CGRP, a potent vasodilator, is released from intracranial afferents during migraine attacks. This vasodilatory effect of CGRP is mediated by its action on CGRP receptors, which stimulates the adenyl cyclase and increases cyclic adenosine monophosphate (cAMP), thus producing potent vasodilation via the direct relaxation of vascular smooth muscle [32][33][115,116]. In response to prolonged noxious stimuli, SP is released from trigeminal sensory nerve fibers around dural blood vessels, leading to endothelium-dependent vasodilation [34][82]. VIP also contributes to neurogenic inflammation by inducing vasodilation [35][117] (Figure 12B).
1.2. Plasma Protein Extravasation
Another critical feature of neurogenic inflammation is PPE. Based on preclinical studies, the neurogenic PPE plays a role in the pathogenesis of migraine [36][118]. In several studies, following electrical stimulation of the trigeminal neurons or intravenous capsaicin, the peripheral nerve endings in the dural vasculature released SP, which caused plasma protein leakage and vasodilation through the NK-1 receptors [37][119]. Transduction of the SP signal through the NK1 receptor occurs via G protein signaling and the secondary messenger cAMP, ultimately leading to the regulation of ion channels, enzyme activity, and alterations in gene expression [38][120]. SP can indirectly influence plasma extravasation by activating mast cell degranulation, which results in histamine release [39][83]. In addition, NKA is able to induce plasma protein efflux and activate inflammatory cells [40][121] (Figure 12C).
1.3. Mast Cell Degranulation
It is well known that dural mast cells play a role in the pathophysiology of migraine [41][122]. Meningeal mast cells are in close association with neurons, especially in the dura, where they can be activated following trigeminal nerve and cervical or sphenopalatine ganglion stimulation [42][123]. The release of neuropeptides, such as CGRP, PACAP, and SP, from meningeal nociceptors can cause the degranulation of mast cells [43][124], resulting in the release of histamine and serotonin and selectively can cause the release of pro-inflammatory cytokines, such as TNF-α, IL-1, and IL-6 [44][45][46][125,126,127]. The plasma and CSF levels of these mediators (e.g., CGRP, TNFα, and IL-1β) are enhanced during migraine attacks [20][104]. VIP promotes degranulation of mast cells [47][128], similar to the effects of SP [39][83]. It was found that CSD can induce intracranial mast cell degranulation and promote the activation of meningeal nociceptors [48][49][129,130]. Besides these, according to several studies, mast cells can be activated by acute stress [42][50][51][123,131,132], which is known to precipitate or exacerbate migraines [52][53][133,134]. Based on these findings, mast cells in themselves may promote a cascade of associated inflammatory events resulting in trigeminovascular activation (Figure 12D).
1.4. Microglia Activation
Microglia appears in the CNS and can exert neuroprotective and neurotoxic effects as well. Under the influence of inflammatory stimuli, microglia can become efficient mobile effector cells [54][135]. Microglia activation leads the production of inflammatory mediators and cytotoxic mediators (e.g., NO, reactive oxygen species, prostaglandins) [55][56][136,137], which might disrupt the integrity of the blood brain barrier, thereby allowing leukocyte migration into the brain [57][138]. Microglia express receptors for neurotransmitters, such as glutamate, gamma- aminobutyric acid, noradrenaline, purines, and dopamine [58][139]. It has been described that activation of ion channels is related to the activation of microglia; therefore, neurotransmitters probably influence microglia function [59][140]. Glutamate leads to neuronal death but is also an activation signal for microglia [60][141]. Activation of glutamate receptors causes the release of TNF-α, which, with microglia-derived Fas ligand, leads to neurotoxicity [61][142]. Besides this, Off signals from neurons appear important in maintaining tissue homeostasis and limiting microglia activity under inflammatory conditions, presumably preventing damage to intact parts of the brain [62][143]. Endothelin B-receptor-mediated regulation of astrocytic activation was reported to improve brain disorders, such as neuropathic pain [63][144]. SP also directly activates microglia and astrocytes and contributes to microglial activation [64][65][145,146], initiating signaling via the nuclear factor kappa B pathway, leading to pro-inflammatory cytokines production [66][147] (Figure 12E).
1.5. Cytokines, Chemokines
Cytokines are small proteins produced by most cells in the body, which possess multiple biologic activities to promote cell-cell interaction [67][148]. There is evidence that cytokines play an important role in several physiological and pathological settings, such as immunology, inflammation, and pain. [68][149]. The most important pro-inflammatory cytokines include IL-1, IL-6, and TNFα, and the key chemokine is IL-8 [68][149]. Cytokines and chemokines are released by neurons, microglia, astrocytes, macrophages, and T cells, and these factors might activate nociceptive neurons [69][150]. TNFα can trigger tissue edema and immune cell infiltration [70][151] and can influence the reactivity of signal nociceptors to the brain and increase blood levels during headaches, playing a crucial role in the genesis of migraine [71][152]. Cytokines are considered to be pain mediators in neurovascular inflammation, which generates migraine pain [72][153]. They can induce sterile inflammation of meningeal blood vessels in migraines [73][154]. Besides this, elevated levels of chemokines can stimulate the activation of trigeminal nerves and the release of vasoactive peptides; thereby, they can induce inflammation [74][155]. Based on these, cytokines and chemokines might contribute to migraine.
2. Current Treatments and Advances in Preclinical Research
Triptans are widely used to relieve migraine attacks; acting as agonists on 5-hydroxytryptamine receptors (5-HT1B/1D), they can cause the constriction of dilated cranial arteries and the inhibition of CGRP release [75][178]. In an animal model of migraine, after electrical stimulation of the TG, sumatriptan attenuates PPE by preventing the release of CGRP [76][179]. In knockout mice and guinea pigs, it has been shown that 5-HT1D receptors have a role in the inhibition of neuropeptide release, thereby modifying the dural neurogenic inflammatory response [77][180]. The use of triptans is limited by their vasoconstrictive properties. As triptans are not effective in everyone, they often lead to medication overuse, triggering migraine to become chronic (Table 12).
Current treatments and advances in preclinical research.
Table 12. Current treatments and advances in preclinical research.
| Drug Class | Drug | Target | FDA Appoved |
|---|---|---|---|
| NSAIDs | Acetylsalicylic acid | COX1–2 | yes |
| Ibuprofen | yes | ||
| Diclofenac potassium | yes | ||
| Paracetamol | yes | ||
| Triptans | Sumatriptan | 5-HT1D receptor | yes |
| Zolmitriptan | yes | ||
| Almotriptan | yes | ||
| Rizatriptan | yes | ||
| Frovatriptan | yes | ||
| Naratriptan | yes | ||
| Eletriptan | 5-HT1B/1D receptor | yes | |
| Ditans | Lasmiditan | 5-HT1F receptor | yes |
| Gepants | Ubrogepant | CGRP receptor | yes |
| Rimegepant | yes | ||
| Atogepant | no | ||
| Vazegepant | no | ||
| Ergot alkaloids | Ergotamine tartrate | α-adrenergic receptor,5-HT receptors | yes |
| CGRP/CGRP receptor monoclonal antibody | Erenumab | CGRP receptor | yes |
| Eptinezumab | CGRP ligand | yes | |
| Fremanezumab | yes | ||
| Galcenezumab | yes | ||
| NK1R antagonists | Aprepitant | NK1 receptor | yes |
| PACAP/PAC1 receptor monoclonal antibody | ALD1910 | PACAP38 | no |
| AMG-301 | PAC1 receptor | no | |
| Endocannabinoids | 2-Arachidonoylglycerol | CB1 receptor | no |
| Anandamide | CB1 receptor | no |
Ditans target the 5-HT1F receptor, which is expressed in the cortex, the hypothalamus, the trigeminal ganglia, the locus coeruleus, the middle cerebral artery, and the upper cervical cord. Lasmiditan is the first drug approved for clinical use. Contrary to triptans, Lasmiditan does not cause vasoconstriction. The activation of 5-HT1F receptor inhibits the release of CGRP and probably SP from the peripheral trigeminal endings of the dura and acts on the trigeminal nucleus caudalis or the thalamus [78][181].
Besides triptans and ditans, acute treatments of migraine headaches, i.e., ergot alkaloids and nonsteroidal anti-inflammatory agents (NSAIDs), may decrease the neurogenic inflammatory response [79][182]. NSAIDs have anti-inflammatory, analgesic, and anti-pyretic properties. Their primary effect is to block the enzyme cyclooxygenase and hence mitigate prostaglandin synthesis from arachidonic acid [80][183]. Both acetaminophen and ibuprofen, which can reduce pain intensity, can also be used in children. Magnesium pretreatment increases the effectiveness of these treatments and reduces the frequency of pain [81][184]. Ergotamine has been recommended to abort migraine attacks by eliminating the constriction of dilated cranial blood vessels and carotid arteriovenous anastomoses, reducing CGRP release from perivascular trigeminal nerve endings, and inhibit the nociceptive transmission on peripheral and central ends of trigeminal sensory nerves [82][185].
An alternative treatment strategy is the use of CGRP-blocking monoclonal antibodies. Monoclonal antibodies have a number of positive properties: (1) a long half-life, (2) long duration of action, and (3) high specificity [83][186]. Four monoclonal antibodies are currently developing for migraine prevention: three against CGRP and one against the CGRP receptor. The safety and tolerability of these antibodies are promising; no clinically significant change in vitals, ECGs, or hepatic enzymes was observed. Blocking of CGRP function by monoclonal antibodies has demonstrated efficacy in the prevention of migraine with minimal side effects in multiple Phase II and III clinical trials [84][187].
Another alternative approach to treating the migraine attack by limiting neurogenic inflammatory vasodilation is the blockade of CGRP receptors by selective antagonists. Gepants were designed to treat acute migraines [85][188]. These bind to CGRP receptors and reverse CGRP-induced vasodilation but were not vasoconstrictors per se [86][189]. Based on these, gepants may be an alternative if triptans are contraindicated. Currently, two gepants (Ubrogepant, Rimegepant) are available on the market, but several are in development.
In gene-knockout studies, the hypothesis the tachykinins are the primary mediators of the PPE component of NI has been strengthened [87][88][190,191]. Following topical application of capsaicin to the ear, the PPE was decreased in Tac1-deficient mice compared to wild-type mice [89][192]. Following activation of the trigeminal system by chemical, mechanical, or electrical stimulation, tachykinin Receptor 1 (TACR1) antagonists seem to be adequate to blocking dural PPE [90][193]. However, lanepitant, a selective TACR1 antagonist, has no significant effect on migraine-associated symptoms [91][194]; moreover, it was found ineffective in a migraine prevention study [92][195]. The only currently available and clinically approved NK1 receptor antagonist is aprepitant, which is used as an antiemetic to chemotherapy-induced nausea in cancer patients [93][196].
In animal models, blockage of TRPV1 receptors was effective to reverse inflammatory pain; however, TRPV1 antagonists produce some serious side effects, e.g., hyperthermia [94][197]. Clinical data suggest that TRPV1 antagonists might be effective as therapeutic options for certain conditions, such as migraine and pain related to several types of diseases. Hopefully, current clinical trials with TRPV1 receptor antagonists and future studies provide an answer as to the role of TRPV1 in inflammatory and neuropathic pain syndromes.
The anti-nociceptive effects of endocannabinoids are thought to be mediated mainly through the activation of cannabinoid receptor type 1 (CB1) [95][198]. Localization of CB1 receptors along the trigeminal tract and trigeminal afferents [96][97][199,200] suggests that the endocannabinoid system can modulate the neurogenic-induced migraine [98][201]. Clinical data suggested that in migraine patients, the endocannabinoid levels are lower [99][100][202,203]. In animal models of migraine, endocannabinoids can reduce neurogenic inflammation. Akerman et al. reported that capsaicin-induced vasodilation is less after intravenous administration of anandamide (AEA); in addition, AEA significantly prevented CGRP- and NO-induced vasodilation in the dura [101][204]. In a previous study, Nagy-Grócz and colleagues observed that NTG and AEA alone or combined treatment of them affects 5-HTT expression, which points out a possible interaction between the serotonergic and endocannabinoid system on the NTG-induced trigeminal activation and sensitization phenomenon, which are essential during migraine attacks [102][205]. These results raise the possibility that the AEA has a CB1 receptor-mediated inhibitory effect on neurogenic vasodilation of trigeminal blood vessels. Based on these, anandamide may be a potential therapeutic target for migraine. Besides these, the presence of CB1 receptors in the brain makes them a target for the treatment of migraine, blocking not only peripheral but also central nociceptive traffic and reducing CSD. CB2 receptors in immune cells may be targeted to reduce the inflammatory component associated with migraine.
PACAP and its G-protein-coupled receptors, pituitary adenylate cyclase 1 receptor (PAC1) and vasoactive intestinal peptide receptor 1/2 (VPAC1/2), are involved in various biological processes. Activation of PACAP receptors has an essential role in the pathophysiology of primary headache disorders, and PACAP plays an excitatory role in migraine [103][206]. There are two pharmacology options to inhibit PACAP: PAC1 receptor antagonists/antibodies directed against the receptor or antibodies directed against the peptide PACAP [104][207]. Studies of the PAC1 receptor antagonist PACAP (6–38) have proved that antagonism of this receptor may be beneficial even during migraine progression [105][208]. PACAP38 and PAC1 receptor blockade are promising antimigraine therapies, but results from clinical trials are needed to confirm their efficacy and side effect profile.
The tryptophan-kynurenine metabolic pathway (KP) is gaining growing attention in search of potential biomarkers and possible therapeutic targets in various illnesses, including migraine [106][107][209,210]. KYNA is a neuroactive metabolite of the KP, which affects several glutamate receptors, playing a relevant role in pain processing and neuroinflammation [78][181]. KYNA may block the activation of trigeminal neurons, affect the migraine generators, and modulate the generation of CSD [106][108][209,211]. An abnormal decrease or increase in the KYNA level can cause an imbalance in the neurotransmitter systems, and it is associated with several neurodegenerative and neuropsychiatric disorders [109][110][111][112][212,213,214,215]. Based on human and animal data, the KP is downregulated under different headaches; thus, possibly less KYNA is produced [113][216]. It is consistent with the theory of hyperactive NMDA receptors, which play a key role in the development of central sensitization [114][217] and thus in migraine pathophysiology. In an NTG-induced rodent model of migraine, Nagy-Grócz et al. demonstrated a decrease in the expression of KP enzymes after NTG administration in rat TNC [115][218]. Interferons can control the transcription expression of indoleamine 2,3-dioxygenase (IDO), kynurenine 3-monooxygenase (KMO), and kynureninase (KYNU); therefore, the pro-inflammatory cytokines may affect the kynurenine pathway [116][219]. It is difficult for KYNA to cross the blood-brain barrier (BBB); therefore, synthetic KYNA analogs may provide an additional alternative for synthesizing compounds that have neuroprotective effects comparable to KYNA that can cross the BBB effectively. Preclinical studies have shown the effectiveness of KYNA analogs in animal models of dural stimulation [117][118][220,221]. Further preclinical studies are required to understand the role of KYNA analogs in migraine and clinical studies that assess their effectiveness in acute or prophylactic treatment (Figure 24).
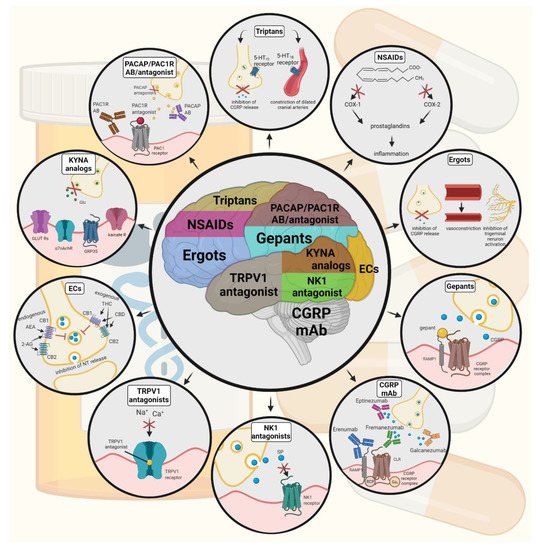
Figure 24. Possible treatments of neurogenic inflammation and migraine. NSAIDs, non-steroidal anti-inflammatory drugs; 5-HT, serotonin; CGRP, calcitonin gene-related peptide; COX, cyclooxygenase; Ab, antibody; NK1, neurokinin 1; TRPV1, transient receptor potential vanilloid receptor; SP, substance P; EC, endocannabinoids; AEA, anandamide; 2-AG, 2-arachidonoylglycerol; CB, cannabinoid receptor; THC, tetrahidrokanabinol; CBD, cannabidiol; NT, neurotransmitter; GLUT R, glutamate receptors; α7AchR, alpha-7 nicotinic receptor; GPR35, G protein-coupled receptor 35; PACAP, pituitary adenylate cyclase-activating polypeptide; PAC1R, pituitary adenylate cyclase 1 receptor.
In animal models of chronic pain and inflammation and several clinical trials, palmitoylethanolamide (PEA), endogenous fatty acid amide, has been influential on various pain states [119][120][121][222,223,224]. In a pilot study, for patients suffering from migraine with aura, ultra-micronized PEA treatment has been shown effective and safe [122][225]. Based on these, PEA is a new therapeutic option in the treatment of pain and inflammatory conditions.