A pneumonia outbreak of unknown origin first emerged in Wuhan, Hubei province of China, during the last months of 2019
1. Introduction
A pneumonia outbreak of unknown origin first emerged in Wuhan, Hubei province of China, during the last months of 2019
. The etiologic agent was identified by the Coronaviridae Study Group of the International Committee on Taxonomy of Viruses (CSG-ICTV) as an unknown positive-strand RNA betacoronavirus (CoV-2)
, the seventh coronavirus known to infect humans, designated as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It emerged as highly transmissible from human to human, and was related to SARS-CoV-1 of 2002 with 82% genome identity. Based on the established practices and studies of phylogeny, it was defined as being of probable zoonotic origin
. The new disease, named the CoronaVIrus Disease 2019 (COVID-19), rapidly spread worldwide due to human-to-human contacts, and the World Health Organization (WHO) declared global pandemic status in March 2020. There is high variability in disease severity, with asymptomatic and paucisymptomatic cases of COVID-19, while severe cases can evolve towards a life-threatening SARS. CoV-2 showed a death rate lower than that of CoV-1, although with pronounced geographical variability
. This suggests that the molecular interactions between the host receptor and the coronavirus variants play an important role in successful infection. However, different host proteins play the roles of receptors/interactors in coronavirus infection, with consequences in comorbidities
.
Recent researches on infectious capacity of this novel coronavirus established that human angiotensin-converting enzyme 2 (hACE2), a homodimer protein attached to cell membrane and exposed at the external environment, is a host receptor for CoV-2 as for CoV-1 in 2002
. Also known as ACEH (ACE homologue), hACE2 belongs to the angiotensin-converting enzyme family of peptidyl dipeptidases (zinc-dependent metalloprotease) and it is homologous to human angiotensin-converting enzyme 1 (hACE1), but with a broader substrate specificity. While hACE1 generates the vasoconstrictor peptide angiotensin 2 (AT2), hACE2 can lower blood pressure by catalyzing the cleavage of angiotensin 1 (AT1) (inactive decapeptide precursor of AT2) into AT1–9 (vasodilator), and AT2 (vasoconstrictor octapeptide) into the vasodilator AT1–7
. Both hACE1 and hACE2 are AT1-converting enzymes and regulators of blood pressure that have counterbalance roles by acting on vasoactive peptides from the renin-angiotensin-aldosterone system (RAAS). In the bloodstream, the renin protease secreted from the kidneys cleaves the angiotensinogen (AGT) secreted by the liver to form AT1, which in turn generates AT2 or AT1–9 from the actions of hACE1 and hACE2, respectively. AT2 is cleaved by multiple enzymes, most importantly hACE2, to form AT1–7. AT2 also forms AT4 via the action of aminopeptidases (APs), both AT2 and AT4 act via AT1 receptors. As hACE2 opposes the actions of AT2, it has a key role in the RAAS. Therefore, there is a beneficial effect in hypertension and cardiovascular diseases when AT2 concentration decreases.
Although hACE2 is hijacked by some coronaviruses, its primary physiological role is in the hydrolysis of AT1 and AT2, peptide hormones that control vasoconstriction and blood pressure
. It is a type I membrane protein primarily expressed in the lungs, heart, kidneys, liver, and small intestine, while its decreased expression is associated with cardiovascular diseases. The monomer of hACE2 consists of an N-terminal peptidase domain (PD) and a C-terminal collectrin-like domain that ends with a transmembrane alpha helix and an approximately 40-residue intracellular segment. Collectrin is a renal protein without catalytic domain and has no similarity with hACE1. Thus, hACE2 may have evolved as a chimera between the ACE-like domain and the collectrin domain. The PD of hACE2 is responsible for processing AT1 to produce AT1–9, which is then processed by other enzymes to become AT1–7, or directly to cleave AT2 to give AT1–7. In contrast with hACE1, hACE2 does not hydrolyze bradykinin and is not inhibited by hACE1 inhibitors (ACE-I). At the onset of the pandemic, the use over time of ACE-I has been postulated to increase susceptibility to COVID-19 in patients with hypertension
[24]. RAAS-inhibition can upregulate the expression of the hACE2 receptor, so the therapeutic treatment with ACE-I might promote the infection. However, to date, no evidence exists that RAAS inhibitors can increase the host’s susceptibility to COVID-19.
The trimeric glycoprotein spike of coronaviruses is anchored in the envelope of the characteristic virion, and many copies of these macromolecules give the likeness of a crown, “corona” in Latin, the word used for the name of these viruses. The spike mediates the recognition of the host hACE2 receptor throughout its receptor-binding domain (RBD), corresponding to the 318–510 sequence region of CoV-2. One of the three spike chains exposes RBD in a structural conformation easy to reach by the hACE2 receptor. So, the spike directly binds with its RBD to the host receptor hACE2-PD, exploiting hACE2 to carry on the infection of the human host (Figure 1).
The specific amino acid sequence of this spike portion plays a key role in conferring to CoV-2 the ability to infect humans, being known as responsible for the species specificity
. RAAS-inhibition can upregulate the expression of the hACE2 receptor, so the therapeutic treatment with ACE-I might promote the infection. However, to date, no evidence exists that RAAS inhibitors can increase the host’s susceptibility to COVID-19.
The trimeric glycoprotein spike of coronaviruses is anchored in the envelope of the characteristic virion, and many copies of these macromolecules give the likeness of a crown, “corona” in Latin, the word used for the name of these viruses. The spike mediates the recognition of the host hACE2 receptor throughout its receptor-binding domain (RBD), corresponding to the 318–510 sequence region of CoV-2. One of the three spike chains exposes RBD in a structural conformation easy to reach by the hACE2 receptor. So, the spike directly binds with its RBD to the host receptor hACE2-PD, exploiting hACE2 to carry on the infection of the human host (Figure 1).
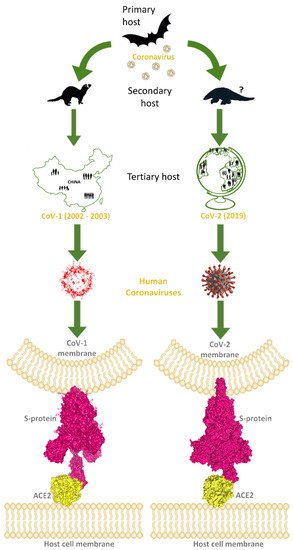
Figure 1. This image explains how coronaviruses spread from bats, as primary hosts, to humans, as tertiary hosts, showing complex spike–ACE2 interactions. Both originated from bats, considered natural reservoirs of SARS-like coronaviruses, CoV-1 and CoV-2 infected the human population via intermediate animal hosts in which viruses underwent an adaptation, exploiting their rapid evolution ability. In the case of CoV-1 (left side of the image), the intermediate host was identified in the masked palm-civets and, by subsequent mutations, CoV-1 became able to jump into humans (tertiary host) and led to the epidemic outbreak that emerged in China and spread in few other countries, to a lower extent, from the end of 2002 to 2003 July. For CoV-2 (right side of the image), the intermediate host is still uncertain, but was probably pangolin, from which CoV-2-related coronaviruses have been isolated. However, after the rapid evolution of these viruses in the secondary hosts, CoV-2 became able to infect the human population causing the COVID pandemic in 2019. Human coronaviruses can interact by the RBD of their trimeric spike glycoproteins, exposed on the membrane of viral envelope, with hACE2 proteins, expressed on the surface of host cells. At the bottom of the image, these interactions are visible for hACE2 and the CoV-1 spike (PDB code: 6CS2; left side, in yellow and magenta surfaces, respectively), and for hACE2 interacting with the CoV-2 spike (PDB codes: 7SBO, 7C8D; right side, in yellow and magenta surfaces, respectively).
The specific amino acid sequence of this spike portion plays a key role in conferring to CoV-2 the ability to infect humans, being known as responsible for the species specificity
. Following infection, the ligand-receptor interaction occurs, the complex spike–ACE2 is formed, and spike is cleaved into two fragments by serine proteases, such as transmembrane protease serine 2 (TMPRSS2): the N-terminal S1 fragment containing the RBD bound to ACE2–PD, and the C-terminal S2 fragment responsible for the membrane fusion of the virus with the host cell
. Therefore, the molecular interaction and the stable binding between spike RBD and ACE2-PD are propaedeutic to the invasion of host cells by CoV-2. The S1 fragment of CoV-2 has a sequence portion with around 70% shared amino acid identity with the corresponding sequence of CoV-1, while the S2 fragment shares 99% identity with the corresponding sequence of CoV-1
. More particularly, inside the S1 fragment, RBD shows high sequence identity between the two coronaviruses with the exception of its C-terminal region, which is involved in the direct binding with the hACE2 receptor
. Therefore, spike RDB is under selective pressure to evade host immune response. This aspect would explain the high mutation frequency observed in spike RBD. Thus, anticoronavirus antibodies (Ab) designed against the core domain of spike RBD and the S2 fragment should be potentially effective with a broad spectrum versus genetic variants of spike
. Novel spikes may likely increase the virulence by evading the host immunity within species and enabling host-switches by altering cross-species receptor recognition. Additionally, the spike has been identified as a critical recombination hotspot
. Therefore, it is very likely that the sequence variability of the spike, as well as of the hACE2 host receptor, may modulate virion intake and consequent disease severity. Consequently, the spike represents an optimal target for the development of RNA/viral vector vaccines, monoclonal Ab, diagnostics, and therapies
. Moreover, spike RBD can be extremely useful for in silico function predictions based on structure, also as a result of mutations, in the interactions with neutralizing Ab and hACE2
.
CoV-2 is being spread much more rapidly than CoV-1. Although many studies have been carried out, it has not yet been definitely established whether the interaction of spike–hACE2 is stronger with CoV-1 or CoV-2, and how this aspect can be put in relation with their different infectivity levels. The computational studies in the literature seem to report conflicting results. Preliminary in silico analysis predicted the free energy values of binding between the spike RBD (CoV-1 and CoV-2) and hACE2. First molecular docking simulations between the spike RBD of CoV-1 and hACE2 showed that their interaction is energetically favored with respect to CoV-2, which nevertheless showed a significant binding affinity to hACE2
. Further in silico researches by molecular modeling and docking showed low binding energy in the CoV-2 spike–ACE2 complex as compared to CoV-1
. Based on the structure of the CoV-2 spike experimentally solved in the prefusion conformation and on the kinetics of this interaction quantified by surface plasmon resonance, another study evidenced that the CoV-2 spike binds to hACE2 with higher affinity (10- to 20-fold) than the spike of CoV-1
. In the same study, many differences at molecular level were evidenced when the spikes of CoV-1 and CoV-2 interacted with hACE2. In this regard, the spike of CoV-2 uses the hACE2 receptor less efficiently than the spike of the CoV-1 strain of 2002, but more efficiently than the CoV-1 strain of 2003
. The CoV-2 mutations located in the spike-RBD region likely cause higher infectivity and lower pathogenicity than CoV-1 of 2002 with around 10% mortality rate
. Although the genome of CoV-2 has 82% nucleotide identity with CoV-1, and the spike of CoV-2 shares about 76% sequence identity with that of CoV-1
, the free energy of binding between the spike and hACE2 is comparable for CoV-2 and CoV-1
. These results are compatible with the fact that the CoV-2 spike-RBD residues at the spike–ACE2 interface may have evolved in extremely complex ways from different common ancestors. Overall, the results of these studies differ on how the structural peculiarities between the two coronaviruses in the binding of spike to hACE2 can differently stabilize the ligand–receptor interactions of the complex spike–ACE2.
Several other proteins have been investigated for the potential activity as receptor or interactor of the spikes of coronaviruses, including CoV-2. TMPRSS2 is a serine protease upregulated by androgen hormones; it is involved in the infection process of many viruses, including coronaviruses, acting on the spike and hACE2, and facilitating virus–cell membrane fusion
. Another receptor/interactor of the spike is neuropilin-1 (NRP1), a cell-surface receptor for vascular endothelial growth factor 165 (VEGF-165) and semaphorins. NRP1 binds furin-cleaved substrates, and is implicated in CoV-2 infectivity
. Other receptors or interactors of coronaviruses, and potentially of CoV-2, are also known. Dipeptidyl peptidase 4 (DDP4) is a glycoprotein membrane receptor involved in T-cell activation, with peptidase activity, and it is known as the MERS receptor
. Another serine protease, TMPRSS11D, cleaves and activates the spike of human coronavirus 229E (HCoV-229E), facilitating its cell entrance
. The C-type lectin domain family 4 member M (CLEC4M) is a membrane protein, known as an attachment receptor for hepatitis C virus (HCV), Ebola virus, human coronavirus 229E, and CoV-1
. Many of these receptors are involved in several pathologies, and more in particular in the main COVID-19 comorbidities, thus suggesting that tissue expression of these proteins may be related to the epidemiological features of COVID-19 patients
. Moreover, several studies have proposed sialic acids on the host cell surface as possible co-receptors, acting as a further attachment mechanism that facilitates CoV-2 to enter the cell
.
To discover potential molecular targets, as there are only a few functional therapeutic agents and several vaccines are currently available, a more in-depth understanding of the molecular interaction mechanisms underlying the initial steps of infection is required. In fact, massive interventions were directed against the first CoV-2 that appeared in 2019, but the emergence of its genetic variants, above all related to spike mutations, presents new challenges based on their high transmissibility and putting in doubt the efficacy of the first vaccines. Therefore, more recent studies have set out to investigate the structural interactions at the chain–chain interface in the spike–ACE2 complexes of crystallographic structures of both CoV-1 and CoV-2 available in the Protein Data Bank (PDB), the single worldwide repository of information about the 3D structural data of biological macromolecules [36]. Structures of the claw-like ACE2-PD in complex with the RBD or spike have revealed the molecular details of their interaction. On the one hand, these studies showed what specific amino acid residues are involved in ligand–receptor binding and how they interact at the spike–ACE2 interface. On the other hand, they allowed to evidence interesting structural characteristics, and differences between CoV-1 and CoV-2. Therapy and prevention of COVID-19 could benefit from these findings, because the spike is a key target for designing therapeutic agents and a viral antigen for optimal vaccine and monoclonal Ab development.
To discover potential molecular targets, as there are only a few functional therapeutic agents and several vaccines are currently available, a more in-depth understanding of the molecular interaction mechanisms underlying the initial steps of infection is required. In fact, massive interventions were directed against the first CoV-2 that appeared in 2019, but the emergence of its genetic variants, above all related to spike mutations, presents new challenges based on their high transmissibility and putting in doubt the efficacy of the first vaccines. Therefore, more recent studies have set out to investigate the structural interactions at the chain–chain interface in the spike–ACE2 complexes of crystallographic structures of both CoV-1 and CoV-2 available in the Protein Data Bank (PDB), the single worldwide repository of information about the 3D structural data of biological macromolecules
2. Spike–ACE2 Interactions
2.1. Analysis of the Interaction of hACE2 with the Spikes from CoV-1 and CoV-2
The amino acid sequences of RBDs from CoV-1 and CoV-2 spikes present approximately 60% of identical amino acids [37]. Sequence differences between CoV-1 and CoV-2 are mainly present in two regions of RBD, i.e., from position 430 to 462 and from position 470 to 503 (CoV-2 numbering). The X-ray crystallographic structures of hACE2 in complex with the CoV-2 or CoV-1 spike have been solved and made available to the scientific community by means of the PDB archive [36]. Structures of the claw-like ACE2-PD in complex with the RBD or spike have revealed the molecular details of their interaction. On the one hand, these studies showed what specific amino acid residues are involved in ligand–receptor binding and how they interact at the spike–ACE2 interface. On the other hand, they allowed to evidence interesting structural characteristics, and differences between CoV-1 and CoV-2. Therapy and prevention of COVID-19 could benefit from these findings, because the spike is a key target for designing therapeutic agents and a viral antigen for optimal vaccine and monoclonal Ab development. . These complex structures have been investigated in detail to find what amino acids are responsible for molecular interaction. It has been shown that 29 and 33 amino acid residues of spikes from CoV-2 and CoV-1 interact with 32 and 33 residues of receptor hACE2, respectively, at least in one of the complex structures [37]. Most of the spike amino acids involved in the binding interface are located in correspondence of the RBD regions differing between the spikes from CoV-2 and CoV-1. In more detail, out of 29 interacting amino acids of the CoV-2 spike, 23 residues interact with the receptor in all investigated complexes (Figure 2). Similarly, out of 33 amino acids of the CoV-1 spike, 23 residues interact with hACE2 in all investigated complexes.
Figure 2. Amino acid residues of the spikes of CoV-2 and CoV-1 interacting with hACE2 at the binding interface, and vice versa (adapted from Giordano et al. 2021 [37]). The amino acids are color-coded: red = negatively charged residues; cyan = positively charged residues. The side arrows indicate the relative positions of the eight amino acid residues conserved to CoV-2 and CoV-1 spikes. The network of interactions between hACE2 and CoV-2/CoV-1 spikes (presented side-by-side) is restricted to H-bond and generic interactions (thin lines) and salt-bridges (large lines).
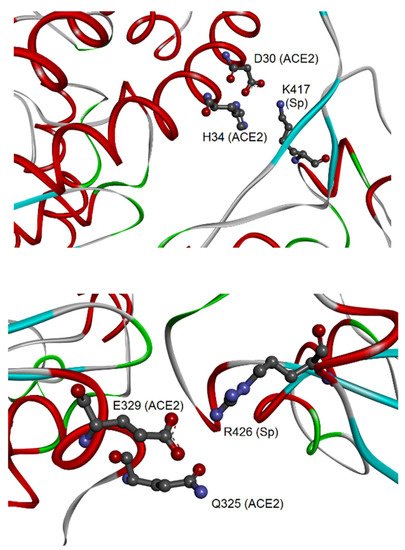
Therefore, these interactions may be particularly relevant for the virus-specific recognition modes of the hACE2 protein. By comparing the two sets of 23 amino acids from CoV-2 and CoV-1, it was possible to identify 19 residues that have a correspondence in the two aligned spike sequences for all analyzed complexes. Out of these residues, eight are identical in both sequences: Y449, Y453, N487, Y489, G496, T500, G502, and Y505 (numbers referred to the CoV-2 spike). These identical residues that interact with the cell receptor in all complexes may have a key function in the binding of CoV-2/CoV-1 spikes with hACE2. Their conservation in the two coronaviruses suggests that functional constraints may have protected them from evolutionary pressure, despite their localization in the two regions of the RBDs with higher sequence variability (position 430–462 and 470–503, see as mentioned above), or very close for Y505. The importance of such observation is related to determining important conserved epitopes of spikes exploitable for maximizing Ab elicitation upon vaccination, above all in the cases of spike variants [26][38]. Nevertheless, the other 15 residues were also notable, being specific to each spike and involved in the interactions of all available spike–ACE2 complexes. Out of 15 residues, there are other 11 amino acids from each spike that, although not identical to each other, correspond in the sequence position and interact with hACE2 (for details, see Table 1 in [37]). In most cases, the conservation of their chemical interactions with hACE2, similarly to what was observed for the eight amino acids already listed, could point out conserved functional features. At last, four amino acid residues of the CoV-2 spike, i.e., K417, G446, G485, and F490, make peculiar interactions with hACE2. The most interesting interaction is related to the residue K417, which binds D30 of hACE2 by H-bond and salt bridge interactions (see Figure 3).
Figure 3. Molecular view of the peculiar interactions formed by CoV-2 K417 and CoV-1 R426 residues of the spike (adapted from Giordano et al., 2021 [37]). The upper panel shows the interaction of CoV-2 spike–K417 with ACE2-D30 and H34; both proteins are represented as backbone ribbons, with helix regions in red, beta-strand regions in cyan, and turns in green. The lower panel shows the interaction of CoV-1 spike–R426 with ACE2-Q325 and E329, with proteins drawn similarly to the upper panel.
This represents an important structural difference in the binding to hACE2 of the CoV-2 spike in comparison to the spike of CoV-1. For this last protein, the four peculiar residues that interact with hACE2 are Y481, R426, T485, and Q492. Also in this case, one of them, i.e., the residue R426, forms an H-bond and salt bridge with one residue of hACE2, i.e., E329, and in addition an H-bond with Q325. The discriminative target residues on hACE2 for the interaction of K417 of the CoV-2 spike and R426 of the CoV-1 spike suggest different modes of binding to the receptor in the two coronaviruses.
2.2. Different Regions of hACE2 Interact with Spikes of CoV-1 and CoV-2
While hACE2 has a negatively charged binding surface, spike RBDs are overall positively charged, forming the complex spike–ACE2 by attractive forces [39]. This is also in agreement with the observation that the spike RBD is recognized mainly by polar amino acid residues located into the extracellular PD of hACE2 [20][21]. At the same time, peculiar different amino acid residues and salt bridge/H-bond positions utilized by the spikes of CoV-1 and CoV-2 allow them to bind differently with hACE2 [39]. The binding interface between hACE2 and CoV-2 spike has been investigated by experimental methods since the first half of 2020, and three models of complexes were deposited in the PDB archives. These structures have been studied by several authors in order to analyze in detail the molecular interaction of the spike with the human protein recognized as the main cellular receptor. Looking at the interface from the hACE2 side, 33 amino acid residues interact with CoV-1 and 32 residues with CoV-2, respectively (as mentioned above), with 30 of them being in common.
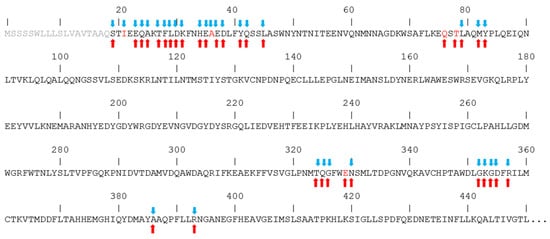
In Figure 4, the sequence of the hACE2 region that interacts with the spike is shown, putting in evidence the amino acids that interact with spikes from both CoV-1 and CoV-2. In fact, despite the high similarity between the spikes of the two coronaviruses, it has been reported that they interact with hACE2 by creating peculiar interactions, although in the context of similar structures. In particular, the unique salt bridge observed with the spike of CoV-2 involves hACE2 D30, while the unique salt bridge observed with the spike of CoV-1 involves hACE2 E329. Similarly, a peculiar H-bonds network binds the spike from CoV-2 to the N-terminal region of hACE2, while in the case of the spike from CoV-1 a distinctive network is formed with the central region of the hACE2 sequence. The amino acids of hACE2 that interact only with the CoV-2 spike are I21 and A36, while Q76, T78, and E329 interact only with the CoV-1 spike.
Figure 4. Interaction of hACE2 amino acids with the spikes of CoV-2 (blue arrows) and CoV-1 (red arrows). Amino acid letters in red indicate unique interaction. Segment 1–18 of the sequence is shown in grey because it is not present in the experimental structures.
Further surface interactions concern hydrophobic contacts as reported in the literature (see Figure 12 of [12] and Supplementary Table S1 of [37]). In particular evidence, hydrophobic interactions are formed by two tyrosine residues conserved in both coronaviruses, i.e., Y489 and Y505 of CoV-2, corresponding to Y475 and Y491 of CoV-1. The conservation of these two tyrosines suggests that their aromatic rings may represent important interaction points with hACE2. It is also interesting to note the peculiar presence of the aromatic rings of F456 and F486 at the surface in the case of CoV-2, corresponding in CoV-1 to L443 and L472, respectively. These two phenylalanine residues of CoV-2 form stacking interactions with the aromatic residues F28 and Y83 at the ACE2 surface, whereas in the case of CoV-1 the two leucine residues have also hydrophobic contacts with ACE2 residues, but evidently do not give rise to aromatic interactions.
The analysis of the complex binding network on the ligand–receptor surfaces revealed that both the spikes interact with hACE2 in correspondence with the N-terminal region (residues 19–83) and central sequence region (residues 324–393). The N-terminal region forms discriminative interactions with CoV-2, as the salt bridge between D30 of hACE2 and K417 of spike. The central region, which also includes the residues 345–346 responsible for the substrate binding [40], has peculiar interactions with CoV-1, as the H-bonds and salt bridge of hACE2 Q325 and E329 with R426 of the spike. These differences observed at the surface of the interaction with the hACE2 receptor show that the two spikes differentially bind to the two districts on the surface of hACE2, with potential consequences for the mechanisms of infection and virulence.
2.3. Structural Predictions of CoV-2 Mutations on the Spike–ACE2 Binding
The modulation of the interactions of the spike on the two surface regions of hACE2 could be related to the different infection ability, also including the CoV-2 variants. A structural comparison of the amino acids at the chain–chain interface can be used to design solutions effective against the binding to the receptor hACE2 of the CoV-2 spike and its mutational variants.
With regard to the mutations of the spike RBD, four CoV-2 major lineages were identified [41][42][43]: Alpha (B.1.1.7) with the mutation N501Y, first detected in the UK; Beta (B.1.351, in South Africa) and Gamma (P.1, in Brazil) with the three identical mutations K417N, E484K, and N501Y (as in Alpha); and Delta (B.1.617, in India) with the mutations L452R and T478K. Among the spike mutational events, K417 was found to be critical in the binding to hACE2. K417 binds to D30 of hACE2 by salt bridge and H-bond interactions, so the K417N mutation might impair the bond for hACE2 of the Beta and Gamma variants by loss of the positively charged K417. Contrarily, spike N501 interacts with hACE2 by the residue Y41 by H-bond (as the later-mentioned T487 of the CoV-1 spike), and the N501Y mutation in the Alpha, Beta, and Gamma variants, might both maintain the H-bond and add an aromatic interaction by introducing a tyrosine side chain, thus reinforcing the binding to hACE2. However, the sterical hindrance of the tyrosine might introduce perturbations at the chain–chain interface. In fact, recently, simulations of molecular dynamics performed on this mutation showed a reduced binding affinity of the B.1.1.7 spike for hACE2, as the residue Y501 restructured the chain–chain interface conformation of the spike–ACE2 complex by decreasing the linear interaction energy between RBD and hACE2 [44]. It is important to note that the other CoV-2 spike substitutions, i.e., E484K, L452R, and T478K, could have been selected—as these sites include amino acids that are not involved in relevant interactions in wild-type coronavirus. Interestingly, these three mutations introduce three further positive charges on the spike, with a possible gain of charge–charge interaction with the hACE2 surface, in agreement with the investigations by Xie et al. [39] already mentioned above. The gain of additional positive charges on the mutated spikes may create further attractive charge interactions that increase the binding affinity of spike to the hACE2 receptor. However, the double mutation observed in Beta and Gamma variants (i.e., K417N and E484K) removed the interaction couples D30-K417 and K31-E484 based on opposite charges. Anyway, their close spatial proximity on hACE2 suggests that in that region the positive side chain of K417 on the CoV-2 surface might be preserved at least in the opposite-charge couple D30-K484.
In November 2021, a variant of concern named Omicron (B.1.1.529 in South Africa) was classified by the WHO [42]. Although studies on this new variant are rapidly appearing, for now it is difficult to define a clear state of the art. This new variant presents a large number of modifications in the spike. Out of these, about half of amino acid substitutions occur within the RBD and thus may affect the interaction with hACE2 and neutralizing Ab. There are cases in common with previous variants, and novel occurrences. In particular evidence, K417N was also observed in Beta and Gamma variants; S477N in Iota variant; E484A was not observed in previous variants, although E484 was substituted by lysine in many variants; N501Y was also present in Alpha, Beta, and Gamma variants.
However, the precise structural impact of known mutations within the RBD on the binding stability of the spike with hACE2 needs yet to be investigated thoroughly [44].
2.4. Mutagenesis Studies on the Spike–ACE2 Interface from Previous SARS Outbreaks
The first human coronavirus (CoV-1), identified as the etiological agent of atypical pneumonia (successively named SARS), originated in late 2002 in Guangdong Province of China. Within a short time, CoV-1 spread worldwide, affecting more than 8000 individuals and being responsible for over 700 deaths during the first outbreak in 2002–2003 [9]. Infected wildlife animals, traded in the live animal markets in China, were suspected as most likely to be the source of CoV-1 human infections (Figure 1). Masked palm-civets (Paguma larvata Hamilton-Smith, 1827) and raccoon dogs (Nyctereutes procyonoides Gray, 1834) were considered the most important viral carriers, intermediate hosts that boost the spread of the virus from the natural reservoir to humans. In fact, the probable wildlife reservoirs of coronaviruses and SARS-like coronaviruses in China are horseshoe bats (Rhinolophus genus Lacépède, 1799). Coronaviruses were likely subjected to an impressive selective pressure during animal-to-human transmission. The spike is a key determinant in the coronavirus–host interaction and this is also the case in interspecies transmission. It was shown that a few alterations of amino acid residues in the spike lead to changes in viral susceptibility in animal and human hosts. Spikes from the coronavirus of palm-civets and from two human outbreaks were compared by Li and colleagues [9]. The affinity of the spike for hACE2 was the determinant for viral infectivity and disease severity. The spike from the severe 2002 outbreak bound hACE2 more efficiently than the spike from the mild 2003 outbreak and from the virus of palm-civets. The low affinity of the spike from the virus of palm-civets was complemented by mutagenizing to the earlier severe outbreak the amino acid residues K479N and S487T. Moreover, the double mutation from human coronavirus to palm-civet virus N479/T487→K479/S487 inhibited interaction with hACE2. Two facing residues of positively charged lysine, K479 in the spike from the palm-civet virus and K31 in hACE2 (Figure 4), repulsively interfered with the formation of the complex spike–ACE2. The K479–K31 repulsive effect is absent in the infection of palm-civets, as they have the polar residue T31 in their ACE2. The residues N479 and T487 of the CoV-1 spike correspond to the residues Q493 and N501 of CoV-2 spike, respectively, both identified in a recent in silico analysis as interacting with hACE2 [37]. These last findings confirmed results on CoV-1, suggesting the hypothesis of a key role for Q493 and N501 of CoV-2 spike in the high-affinity binding to hACE2. Indeed, Q493 interacts with the hACE2 residues K31 and H35. In addition, K31 interacts with the residue E484 of the CoV-2 spike and other two residues upstream of Q493 (F490 and L492). In the same way, mutagenesis analysis of position 487 showed the key role of a threonine in increasing the affinity for human and non-human ACE2. It corresponds to the residue N501 of CoV-2, subject to the N501Y mutation in the Alpha, Beta, and Gamma variants. In silico analysis showed that N501 interacts with the residue hACE2 Y41 (similarly present in palm-civet and rat ACE2), which in turn interacts with two residues upstream of N501 (Q498 and T500). Altogether, these findings indicate that variations of the CoV-2 spike in the sites 493 and 501 (479 and 487 in CoV-1 spike) are compatible with the binding of novel coronavirus to the human receptor by the recognition of the K31 and Y41 residues [9][37]. These mutational hotspots may be elements predictive in the risk assessment of new coronavirus variants/lineages [43][44]. The same in silico analysis also evidenced the presence of peculiar interactions by R426 of the CoV-1 spike [37], not mutagenized in that study [9], as well as K417 of CoV-2 spike corresponding to V404 of the CoV-1 spike, unknown at that time. Notably, these interactions concern the CoV-2 spike K417 with hACE2 D30, and the CoV-1 spike R426 with hACE2 E329 (Figure 3). These residues are not conserved in mouse and rat ACE2, where the same positions are occupied by N30 and A/T329 [45]. These facts contribute to explain why both the coronaviruses are unable to hijack murine ACE as a cell receptor.
To better understand the coronavirus spillover from animal species to humans, Li et al. (2005) [9] also carried out mutagenesis studies, creating chimeras of hACE2 with palm-civet and rat ACE2. The low affinity for hACE2 of the spike from the palm-civet virus was complemented by altering hACE2 to the palm-civet counterpart in the residues at the positions 31–40 and K353. Moreover, the alterations to the human counterpart of the residues NFS→MYP (position 82–84) and H353K convert rat ACE2, whose wild type does not bind the CoV-1 spike, to an effective cell receptor. The mutations K31D and Y41A on hACE2 interfered with CoV-1 spike interaction, as well as the mutations D355A and R357A downstream of K353. These results allowed localization of the CoV-1 spike-binding domain above the cleft of the catalytic pocket of hACE2 [9]. The results by Li et al. (2005) [9] can be partially applied to the spike–hACE2 interaction of CoV-2, as shown by in silico analysis of experimental complexes [37]. In fact, the region of hACE2 from position 19 to 45 is also interacting with the CoV-2 spike with more interactions than CoV-1 (Figure 2 and Figure 4). Further interacting regions are located at the sequence position 76–83 of hACE2 with Y83, at 324–330, and at 352–357 with three amino acid residues (K353, D355, and R357) important for the binding to CoV-1 spike but less so to the CoV-2 spike [37]. Moreover, the same in silico analysis identified the residues Q325 and E329 interacting with R426 of the CoV-1 spike. However, the mutagenesis of Q325P and E329T to the corresponding residues of rat ACE2 did not show the effects on its binding to the CoV-1 spike [9]. Since the CoV-1 spike has more interactions than CoV-2 within the hACE2 portion 325–357, these could balance the mutagenesis and justify its limited effect.
3. Conclusions and Prospects
Different interactions of the spikes from CoV-1 and CoV-2 with hACE2 can be responsible for the high spread from human to human of the novel coronavirus. The molecular interactions at the chain–chain interface of the spike–ACE2 complex are extremely decisive for the coronavirus attack to human cells. The understanding of the underpinning mechanism of action as well as possible interaction differences, as in the case of spike variants, are essential for developing innovative strategies to fight the pandemic. The in silico analysis on the experimentally determined spike–ACE2 complexes defined which and how residues interact at the ligand–receptor interface at molecular level. These studies evidenced peculiar differences between CoV-1 and CoV-2 with potential health implications. The spike from CoV-2 binds to the hACE2 differently from the spike of CoV-1, and this may contribute to the different contagiousness, regardless of high or low molecular affinity. The complex network of the interactions at the binding interface between hACE2 and the spike showed strong interactions of CoV-2 spike with the N-terminal region of hACE2, while the CoV-1 spike mainly had interactions with the central region of hACE2. Further studies are still needed to unveil the detailed molecular basis of virus–cell recognition, consequent cell invasion, cell–cell transmission, human-to-human spread, and the pathogenesis of CoV-2, as well as to optimize diagnostic, antiviral, and vaccination strategies. Unfortunately, coronaviruses are an example of the evolution of animal viruses into deadly human pathogens. From now on, it is of paramount importance that we watch over wildlife as a strategy for the prevention and control for infectious diseases.