Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xiaotian Zhong and Version 2 by Bruce Ren.
Glycans as sugar polymers are important metabolic, structural, and physiological regulators for cellular and biological functions. They are often classified as critical quality attributes to antibodies and recombinant fusion proteins, given their impacts on the efficacy and safety of biologics drugs
- glycosylation pathways
- N-acetyl-galactosamine
- mannose-6-phosphate
- lysosomal degradation
- Fab glycans
- antibody diversification
- sialylation
- glycome
- O-linked glycans
- therapeutic proteins
1. Introduction
Glycan modification [1][2][3][4][1,2,3,4], in mammalian glycoproteins, glycolipids and recently in RNAs [5], represents the most complex and diverse networks and pathways for post-translational modifications. The tremendous structural diversity of glycan polymers are synthesized without a template, but rather through a sequential step addition by compartmentally restricted cellular glycosylation machineries which employ around 700 genes encoding glycotransferase enzymes, transporters and chaperones required for establishing the ensemble of glycans [1][6][1,6]. Apart from the non-enzymatic glycation between glucose and lysine/arginine [7] as well as cytosolic and nuclear O-GlcNAcylation [8], protein glycosylation processes involve sequentially orchestrated modification reactions in the metabolic networks of the endoplasmic reticulum (ER) and the Golgi during protein trafficking. It has been estimated that the known glycome and the glycosylation network are generated through 16 distinct glycosylation pathways according to sugar–protein linkages, initial monosaccharides linked to proteins, and unique initiating enzymes [1].
Glycan attachments to protein are generally classified into four major types. N-linked glycosylation is through asparagine (Asn) that is initiated at the ER by the en bloc transfer of core glycans via oligosaccharyltransferase (OST) and further modified by various glycoenzymes and glycotransferases in the ER and the Golgi [1][2][3][9][1,2,3,9]. O-linked glycosylation involves covalent modification to the hydroxyl groups of serine (Ser), threonine (Thr), or tyrosine (Tyr) with direct attachments of seven different sugars including N-acetyl-galactosamine (GalNAc), L-fucose (Fuc), N-acetyl-D-glucosamine (GlcNAc), D-mannose (Man), D-glucose (Glc), D-xylose (Xyl), and D-galactose (Gal). GalNAc-type and Xyl-type O-linked glycosylation start at the Golgi by polypeptide GalNAc transferases (GALNTs) and O-xyltransferases (XYLTs), respectively. Fuc, Glc, and GlcNAc types of O-linked glycosylation are initiated in the ER. Mammalian Man-type O-linked glycosylation is initiated in the ER and further modified in the Golgi. The other two ways for glycan attachments are glypiation through GPI linkage and C-linked to tryptophan (Trp) [1].
The natural building blocks for glycans in mammals are 10 monosaccharides including D-glucuronic acid (GlcA), D-ribose (Rib), Fuc, Glc, GlcNAc, Gal, GalNAc, Man, N-acetylneuraminic acid (Neu5Ac), and Xyl, which can be derived from the corresponding dolichol-linked donors or activated donor sugar nucleotides [1][2][3][1,2,3]. The structural diversification of glycans through the sequential addition of monosaccharides mostly occur in the Golgi for oligosaccharide extending, branching, and capping. The final glycan structures are determined by glycosyltransferases’ kinetic properties, their compartmental distributions along the sequential biosynthetic routes, as well as factors such as substrate availability and actions of protein chaperones and glycosidases.
Therapeutic proteins, such as antibodies and recombinant fusion proteins, are glycoproteins in which glycan modifications are often considered critical quality attributes and can be engineered for therapeutic efficacy and safety improvements (according to several reviews [6][10][11][12][13][6,10,11,12,13]). With a global view on the human glycome being established and a deeper understanding on glycosylation pathways, new opportunities in harnessing human protein glycosylation functions are emerging (Figure 1). This article highlights new applications of GalNAc and mannose-6-phosphate (M6P) glycan modification in protein therapeutics (Figure 2). New findings on antibody repertoire glycan diversification, O-linked mannosylation, glycan remodeling on branching, sialylation, and fucosylation were also discussed.
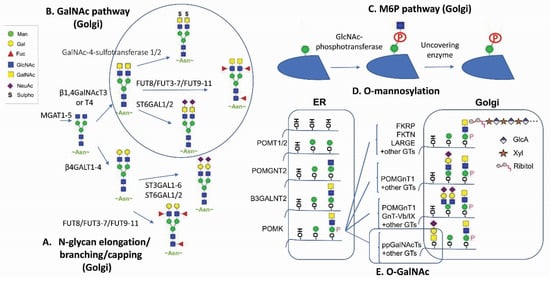
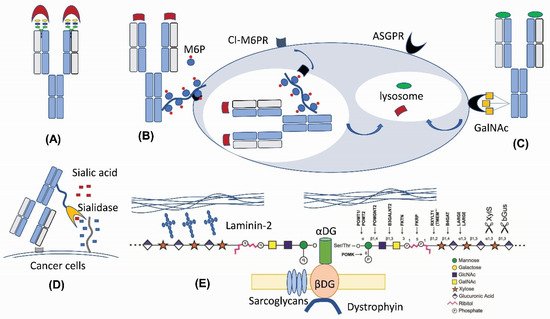
Figure 2. New therapeutic applications of N-linked and O-linked glycan modifications. (A) Fab N-glycan for the antibody diversity [17]. (B) M6P-mediated lysosomal degradation [18]. (C) GalNAc-mediated lysosomal degradation [19][20][21][19,20,21]. (D) Antibody–sialidase fusions or conjugates [22][23][22,23]. (E) O-mannosylation matriglycan as a functional decoration for α-Dystroglycan [24][25][24,25].
2. Glycans as an Unconventional Strategy for Antibody Diversification
N-linked glycans are present in 15–25% of human IgG antibodies’ variable domain (heavy chain variable domain (VH) or light chain variable domain (VL)) regions [26][27][26,27]. These N-glycosylation sites encoded by the V-region genes (so-called Fab N-glycans) are a result of somatic hypermutation [26][28][29][26,28,29], because very few germline alleles carry N-glycosylation consensus sequences (NXS/T) [30]. In recent years, more and more evidences indicate that Fab N-glycans can influence antibody binding affinity. Several mechanisms on how N-glycan in antibody V-regions impacts epitope binding have been proposed, including the bulk size of N-glycan to fill out the space between the antigen epitope and the antibody paratope [31], charge–charge interaction between N-glycan sialic acids and the antigen [17][28][17,28], and through steric hinderance effects that affect the binding [32]. The IgG4 subclass has the highest prevalence of V-region glycosylation (44% versus 11%–15% in other subclasses) [28]. IgE has a two-fold higher propensity for Fab glycans than IgA or IgG1, suggesting that elevated Fab glycosylation might be a hallmark of Th2-like responses [33]. A large portion of autoantibodies in rheumatoid arthritis and certain B-cell lymphomas were found to contain Fab N-glycans [34][35][36][34,35,36], which are also present in human anti-idiotype autoantibodies to adalimumab and infliximab [28]. Removing N-glycans from the complementarity-determining regions (CDRs) of antibodies can lead to a significant decrease in the antibody binding affinity [28][37][38][28,37,38]. Removing N-glycan located within the antigen-binding sites of a human IgG alloantibody decreases its neutralization towards factor VIII (FVIII) procoagulant activity without losing its binding affinity, suggesting that its Fab glycan blocks the interaction between FVIII and the chaperone partner through steric hinderance [32]. Fab glycans in the framework or constant regions play additional roles in increasing antibody stability [29] and in vivo half-life [39].
The structure of N-glycans within the V region are different from those rigid under-sialylated biantennary Fc-glycans attached to Asn297 in the Fc region, because they are typically surface-exposed α2,6-linked sialylated complex biantennary glycans [37][40][41][37,40,41]. The negatively charged sialic acid on these V-region glycans have been found to contribute to the increased binding affinity [28][38][40][28,38,40]. This data indicate that the introduction of N-linked glycans to variable domains is an additional layer for immune repertoire diversification [17].
Engineering N-glycans into antibody-binding sites has been utilized for therapeutic rational design (Figure 2A). Engineering an N-linked consensus site into an ibalizumab light chain recognizes human immunodeficiency virus (HIV)’s envelope glycoprotein gp120 with a loss of an N-glycan in the V5 loop, which is otherwise resistant to the HIV-1-neutralizing activity [31]. Similarly, introducing Fab glycans into adalimumab enhances the TNFα binding of two antibody glycovariants by two-fold [28]. Introducing Fab N-glycans can be a way to decrease antigen-binding poly-reactivity and self-reactivity [42][43][42,43]. The introduction of an N-linked glycan into an antibody-variable domain also has been employed for improving antibody solubility [44][45][44,45]. Although engineering in Fab N-glycosylation can increase manufacturing challenges, the high degree of conformational dynamics from glycans can enhance the chemical diversity of antibody paratopes and thus the functionalities.
3. GalNAc Binder—A New Application Based on Previous Findings
Therapeutic antibodies can exert biological actions on signal transduction pathways by blocking interactions between receptors and ligands. Three recent studies reported a new therapeutic mechanism by conjugating tri-GalNAc to antibodies for directing the lysosomal degradation of several therapeutic ligands and receptors [19][20][21][19,20,21]. These so-called lysosome-targeting chimeras (LYTACs) are capable of inducing a rapid internalization and degradation of membrane targets and soluble targets based on the binding to the liver-specific asialoglycoprotein receptor (ASGPR) and lysosome machineries (Figure 2C).
Naturally, GalNAc residues (Figure 1B,E) can be added to proteins through either N-glycan LacdiNAc modification or O-linked GalNAc addition [1][3][6][1,3,6]. The tight binding to ASGPR or mannose receptor (MR) has been reported for N-glycan LacdiNAc modified proteins [46]. LacdiNAc is a less common N-linked glycan structure [14][47][48][49][50][14,47,48,49,50]. It contains the unique GalNAcβ1-4GlcNAcβ unit, which can be additionally sulfated, fucosylated and sialylated. About 12 glycoproteins are confirmed with N-linked LacdiNAc glycans, i.e., luteinizing hormone. β1,4GalNAcT3 [51] and β1,4GalNAcT4 [52] account for GalNAc transfer and have a broad tissue expression coverage including fetal kidney and brain. It has been reported that adding a carboxyl-terminal 19-amino-acid α-helix stretch with several basic amino acids is sufficient to mediate GalNAc transfer to N-linked oligosaccharides [47][53][54][47,53,54]. Other GalNAc motifs involve three structural loops with aromatic side chains [50], as well as additional unidentified motifs [54].
Since LYTAC molecules conjugated with GalNAc can be targeted for lysosomal degradation, fusing the GalNAc transfer motif to the termini of antibodies or therapeutic fusion proteins should enable the LacdiNAc modification on these proteins during mammalian cell culturing. HEK293 cells express the key glycoenzymes of β1,4GalNAcT3, β1,4GalNAcT4, and GalNAc-4-sulfotransferases-1 and -2, and a stable production in HEK293 has generated clinical and commercial biotherapeutics [55]. In fact, because CHO cells either lack or do not express several glycosyltransferases, therapeutic proteins such as recombinant human erythropoietin are found to be LacdiNAc-modified when expressed in HEK293 cells, but not in CHO cells [55][56][55,56]. Alternatively, CHO cells with necessary glycoenzymes can be engineered for LacdiNAc modification [47].
4. M6P—A Lysosomal Route for Non-Lysosomal Enzymes
M6P modification (Figure 1C) in specific N-linked glycans serves as a recognition signal for lysosomal routing [11][15][57][11,15,57]. When lysosomal hydrolases synthesized in the ER are transported to the cis-Golgi network, they are selectively modified by a two-step reaction. GlcNAc-1-phosphotransferase transfers a GlcNAc-1-phosphate residue from UDP-GlcNAc to C6-positions of specific mannoses in high-mannose N-glycans of lysosomal hydrolases. The GlcNAc-1-phosphotransferase is a Golgi hexameric transmembrane enzyme encoded by two different genes, i.e., GNPTAB and GNPTG [15]. Defects in this key enzyme causes lysosomal storage disease mucolipidosis II and III. The second step of M6P generation is catalyzed by an N-acetylglucosamine-1-phosphodiester α-N-acetyl-glucosaminidase (also known as “uncovering enzyme”) for the removal of the terminal GlcNAc to expose the signal. The uncovering enzyme is a tetrameric type I membrane protein cycling between the trans-Golgi network and the plasma membrane. No pathological conditions are associated with the loss of its enzymatic activity.
In the trans-Golgi network, the M6P modification allows for the segregation of lysosomal hydrolases from other trafficking proteins through a selective binding to two M6P receptors, i.e., the cation-independent M6P receptor (CI-MPR) and/or the cation-dependent M6P receptor (CD-MPR) [15][57][15,57]. The clathrin-coated ligand receptor complex transport vesicles bud off and fuse with late endosomes. At the low pH of the late endosome (Figure 2B), the M6P receptors dissociate from the ligands and be recycled back to the trans Golgi network.
The therapeutic application of M6P modification is the lysosomal delivery of the enzyme replacement therapy for lysosomal diseases, such as Fabry disease, mucopolysacharidosis I, II, and VI, and Pompe disease [58]. High-affinity M6P analogues with good stability, such as mannose-6-phosphonate (M6Pn), could be synthesized [59] and conjugated, like the M6P-containing oligosaccharides [60], to recombinant enzymes for decreasing the effective dose for less accessible tissues. M6P is also present in glycoprotein D of herpes simplex virus (HSV) for virus entry into cells [61]. Recombinant CI-M6PR and pentamannose-phosphate are used to block HSV plaque formation [62]. Most recently, M6P has been exploited for lysosomal degradation [18]. Because the 6-phosphoester of M6P can undergo hydrolysis in human serum, the phosphatase-inert serine-O-M6Pn glycopeptide is conjugated to an antibody to form a different kind of LYTACs that interacts with CI-M6PR for shuttling to the lysosomal compartment for the degradation of extracellular proteins engaged by the antibody component of the conjugates. For the biological production of M6P-modified glycoproteins, one strategy is to utilize engineered yeast cells to synthesize Man-P-6-Man glycans, in which phosphate-capped Man residues can be subsequently removed by a newly discovered α-mannosidase to generate M6P-modified human lysosomal enzymes [63]. These new tools and new findings should enable further glycoengineering of next-generation biologics for lysosomal targeting.