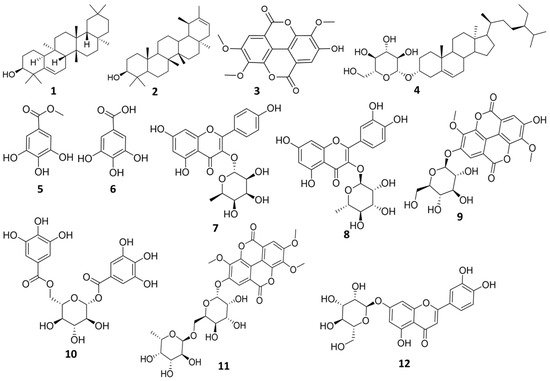
Human African trypanosomiasis is an endemic infectious disease caused by Trypanosoma brucei via the bite of tsetse-fly. Most of the drugs used for the treatment, e.g., Suramin, have shown several problems, including the high level of toxicity. A In our previous study on the anti-trypanosomal potential of Euphorbia species, Euphorbia abyssinica displayed significant anti-trypanosomal activity. Therefore, a phytochemical investigation of the methanolic extract of E. abyssinica was carried out. Twelve compounds, including two triterpenes (1, 2); one sterol-glucoside (4); three ellagic acid derivatives (3, 9, 11); three gallic acid derivatives (5, 6, 10); and three flavonoids (7, 8, 12), were isolated. Compound (10) was obtained for the first time from genus Euphorbia while all other compounds except compound (4), were firstly reported in E. abyssinica. Consequently, an in silico study was used to estimate the anti-trypanosomal activity of the isolated compounds. Several compounds displayed interesting activity where 1,6-di-O-galloyl-d-glucose (10) appeared as the most potent inhibitor of trypanosomal phosphofructokinase (PFK). Moreover, molecular dynamics (MD) simulations and ADMET calculations were performed for 1,6-di-O-galloyl-d-glucose. In conclusion, 1,6-di-O-galloyl-d-glucose revealed high binding free energy, desirable molecular dynamics, and pharmacokinetic properties; therefore, it could be suggested for further in vitro and in vivo studies for trypanosomiasis.
1. Introduction
Trypanosoma brucei is the causative agent of human African trypanosomiasis (HAT), sleeping sickness, via the bite of the tsetse fly. One of the most druggable target enzymes for the treatment of HAT is the trypanosomal phosphofructokinase (PFK) enzyme
[1]. Suramin, a classic PFK inhibitor, is known to exhibit significant side effects such as hypersensitivity, agranulocytosis, and nephrotoxicity
[2].
E. abyssinica J.F. Gmel latex was reported to yield ingenol esters and lathyrane derivatives as minor components besides euphol, euphorbol, lupeol, oleanolic acid, β-sitosterol, and β-sitosterol-3-
O-glucoside. Also, it has shown significant antifungal activity against
Aspergillus flavus, Aspergillus niger, and
Candida albicans [3]. Furthermore,
E. abyssinica has exhibited cytotoxic activity against Caco2 (IC
50 11.3 µg/mL)
[4]. Moreover, the root of
E. abyssinica has shown potent chemosuppressive antimalarial activity against
Plasmodium berghei infection in mice
[5].
Recently, molecular docking is important in the estimation of the bioactivity of chemical compounds against a target and has shown great progress
[6]. The evaluation of drug design depends on the identification and characterization of small-molecule binding sites on the target proteins
[7].
In our previous research, the anti-trypanosomal activity of the methanolic extract of
E. abyssinica against
T. brucei brucei strain TC221 was investigated, and IC
50 values were determined as 17.3 and 19.4 μg/mL after 48 and 72 h incubation, respectively
[8]. Consequently, the current study discusses the molecular modeling study of the compounds isolated from
E. abyssinica J.F. Gmel. against the target proteins (PFK) of
T. brucei. Furthermore, the molecular dynamics and pharmacokinetic properties of the most active compound are also presented in order to conclude the compound activity.
2. Investigation of Methylene Chloride Fraction of E. abyssinica J.F. Gmel
Chromatographic investigation of methylene chloride fraction led to isolation of four compounds. The structure of the isolated compounds was elucidated using 1D NMR and LC-HRMS. Compound (
1): White needle powder (15 mg), m.p. 208–212 °C, gave a positive Libermann–Burchard’s test indicating its steroidal or triterpenoidal nature. LC-HRMS [M + H]
+ m/
z: 427.3931,
Rt: 26.12 calculated for C
30H
50O.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in CD
3OD.
Compound (
2): White crystal powder (25 mg), m.p. 282–285 °C, gave a positive Libermann–Burchard’s test indicating its steroidal or triterpenoidal nature. LC-HRMS [M + H]
− m/
z: 425.3859,
Rt: 28.97 calculated for C
30H
50O.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in CDCl
3.
Compound (
3): Yellowish white amorphous powder (50 mg), m.p. 289–291 °C, produced a positive reaction to FeCl
3 reagent. LC-HRMS [M + H]
+ m/
z: 345.0602,
Rt: 16.99 calculated for C
17H
12O
8.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in DMSO
-d6.
Compound (
4): White amorphous powder (24 mg), m.p. 290 °C; the color of the spot was invisible in TLC and under UV but after spraying with
p-anisaldehyde, it was violet. It gave a positive Libermann–Burchard’s test indicating its steroidal or triterpenoidal nature and gave a positive with Molish’s test indicating its glycosidic nature. LC-HRMS [M + H]
− m/
z: 575.3155,
Rt: 25.82 calculated for C
35H
60O
6.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in DMSO-
d6.
3. Investigation of Ethyl Acetate Fraction of E. abyssinica J.F. Gmel
Chromatographic investigation of ethyl acetate fraction led to the isolation of seven compounds. The structure of the isolated compounds was elucidated using 1D, 2D NMR, and LC-HRMS. Compound (
5): White crystalline powder (17 mg), m.p. 198–200 °C, produced a positive reaction to FeCl
3 reagent. LC-HRMS [M + H]
− m/
z: 183.0301,
Rt: 8.37 calculated for C
8H
8O
5.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in (CD
3OD).
Compound (
6): White crystalline powder (14 mg), m.p. 258–263 °C, produced a positive reaction to FeCl
3 reagent. LC-HRMS [M + H]
− m/
z: 169.0143,
Rt: 5.01 calculated for C
7H
6O
5.
1H-NMR (400 MHz) in CD
3OD and DEPT-Q NMR (100 MHz) in (CD
3OD).
Compound (
7): Yellow powder (10 mg), m.p. 172–174 °C, LC-HRMS [M + H]
+ m/
z: 433.1125,
Rt: 12.70 calculated for C
21H
20O
10.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in CD
3OD.
Compound (
8): Yellow amorphous powder (12 mg), m.p. 179–183 °C, LC-HRMS [M + H]
+ m/
z: 449.1074,
Rt: 11.87 calculated for C
21H
20O
11. TLC investigation revealed an orange spot while it showed a deep purple spot under UV light, which became yellow–green when fumed with ammonia vapor but showed dark orange color with
p-anisaldehde indicating its flavonoid-3-
O-substituted nature.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in CD
3OD.
Compound (
9): Yellowish white amorphous powder (25 mg), m.p. 297 °C, produced a positive reaction to FeCl
3 reagent. LC-HRMS [M + H]
+ m/
z: 493.0970,
Rt: 9.57 calculated for C
22H
20O
13.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in DMSO
-d6.
Compound (
10): Off-white amorphous powder (16 mg), m.p. 180–182 °C, produced a positive reaction to FeCl
3 reagent. LC-HRMS [M + H]
+ m/
z: 485.0921,
Rt: 8.29 calculated for C
20H
20O
14.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in CD
3OD.
Compound (
11): Yellowish white amorphous powder (45 mg), m.p. 267–268 °C, produced a positive reaction to FeCl
3 reagent. LC-HRMS [M + H]
+ m/
z: 653.1704,
Rt: 11.93 calculated for C
29H
32O
17.
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in DMSO-
d6.
4. Investigation of n-Butanol Fraction of E. abyssinica J.F. Gmel
Chromatographic investigation of
n-butanol fraction led to the isolation of one compound. The structure of the isolated compound was elucidated using
1H-NMR, and LC-HRMS. Compound (
12): Yellow amorphous powder (6 mg), m.p. 320–330 °C. UV λ
max (MeOH) nm: 225, 258, 347.5, (AlCl
3) 272.5, 297, 331, 421. LC-HRMS [M + H]
+ m/
z: 449.1074,
Rt: 11.87 calculated for (C
21H
20O
11).
1H-NMR (400 MHz) and DEPT-Q NMR (100 MHz) in CD
3OD. All isolated compounds are represented in (
Figure 1).
Figure 1.
Structure of the isolated compounds from
E. abyssinica
.
5. Docking Study for Anti-Trypanosomal Activity
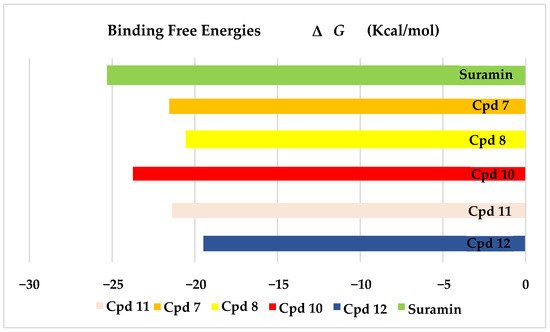
The results of docking procedures contained binding free energies Kcal/mol, binding affinity constant (
ki in nm), distances (in Å) from the main residues, and type of interactions. Notably compounds
10,
7,
11,
8, and
12 in order, showed good binding affinity energies (from −18.9900 to −23.0767 Kcal/mol) when compared to the co-docked ligand suramin as a positive control (
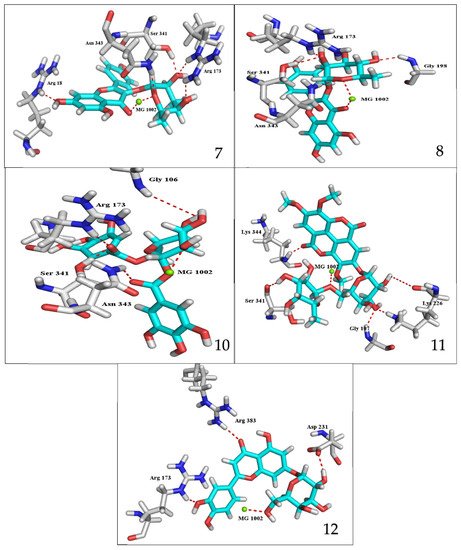
Figure 2). The main residues involved in the interaction between compounds and
T. brucei PFK enzyme were Arg173, Ser341, Asn343, Lys226, Thr201, and Gly107 residues as well as Mg Atom (MG1002) that mark them as good candidates for
T. brucei PFK inhibition, that could be used for the treatment of trypanosomiasis. Hydrogen acceptor and metal interactions were found to be the main formed interactions between compounds and the enzyme. 3D figures of the most active compounds via PyMOL 2.4 software were represented in (
Figure 3).
Figure 2. Binding free energy score of the most active isolated compounds and suramin with T. brucei PFK enzyme (PDB ID:3F5M).
Figure 3.
3D interaction caption of the top docking pose of the most active isolated compounds.
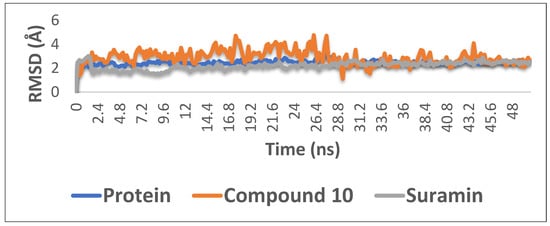
6. Molecular Dynamics Simulations
With the aim of proofing the reliability of molecular docking results, further computational validation was achieved through a number of MDS experiments and binding free energy (ΔG) calculations on compound
10 (1,6-di-
O-galloyl-
d-glucose), as well as suramin. As seen in
Figure 4, compound
10 was able to achieve stable binding inside the enzyme′s (i.e., phosphofructokinase, PDB ID:3F5M) active site with an average RMSD from the initial docking pose of 3.1 Å; however, it showed higher fluctuation in comparison with the standard drug suramin. Accordingly, it obtained a binding free energy value (ΔG) of −7.1 kcal/mol (ΔG of suramin was −8.8 kcal/mol).
Figure 4. The RMSD curve from the molecular dynamics simulations of compound 10. The X-axis represents the simulation time (in ps), while the y-axis represents the RMSD value (in nm).
7. Prediction of the Pharmacokinetic Properties and Toxicological Properties Using ADMET
After the molecular docking studies of 12 isolated compounds, the absorption, distribution, metabolism, elimination, and toxicity (ADMET) of the best dock scored compound along with suramin were evaluated (
Table 1).
Table 1.
ADMET properties of compound
10
and suramin.
| Properties |
Compound 10 |
Suramin |
| Absorption |
Caco-2 permeability (log Papp in 10−6 cm/s) |
−1.682 |
−3.097 |
| HIA (% Absorbed) |
15.64% |
0 |
| P-glycoprotein substrate |
Yes |
Non |
| P-glycoprotein I inhibitor |
Non |
Non |
| P-glycoprotein II inhibitor |
Non |
Non |
| Pure water solubility (log mol/L) |
−2.895 |
−2.892 |
| Skin Permeability (log Kp) |
−2.735 |
−2.735 |
| Distribution |
BBB Permeability (log BB) |
−2.435 |
−4.438 |
| CNS permeability (log PS) |
−4.668 |
−4.991 |
| VDss human (log L/kg) |
1.614 |
−0.007 |
| Fraction unbound human (Fu) |
0.347 |
0.379 |
| Metabolism |
CYP 2C19 inhibitor |
Non |
Non |
| CYP 2C9 inhibitor |
Non |
Non |
| CYP 2D6 inhibitor |
Non |
Non |
| CYP 2D6 substrate |
Non |
Non |
| CYP 3A4 inhibitor |
Non |
Non |
| CYP 3A4 substrate |
Non |
Non |
| CYP 1A2 inhibitor |
Non |
Non |
| Excretion |
Total Clearance (log mL/min/kg) |
0.47 |
−4.065 |
| Renal OCT2 substrate |
Non |
Non |
| Toxicity |
Ames test |
non-mutagen |
non-mutagen |
| Max. tolerated dose human (log mg/kg/day) |
0.49 |
0.438 |
| Oral Rat Acute Toxicity LD50 (mol/kg) |
2.515 |
2.482 |
| Oral Rat Chronic Toxicity LOAEL (log mg/kg-bw/day) |
3.491 |
6.817 |
| hERG I inhibitor |
Non |
Non |
| hERG II inhibitor |
Yes |
Yes |
| T. pyriformis toxicity (log μg/L) |
0.285 |
0.285 |
| minnow toxicity (log mM) |
5.837 |
6.162 |