Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Marco Tutone.
Translational Readthrough-Inducing Drugs (TRIDs) can affect various biomolecular targets, leading to different types of readthrough.
- Cystic Fibrosis
- Non sense mutations
- CFTR rescue
- TRIDs
- Ataluren
- Translarna
- Aminoglycosides
- Oxadiazoles
- ELX-2
- Amlexanox
- Clitocine
1. Aminoglycosides
Aminoglycosides are derivatives of polyamine oligosaccharides commonly used as antibiotics to treat Gram-negative antibacterial infections [36][1]. In fact, they are capable of binding to the site of the bacterial small ribosome unit that generally controls codon-anticodon interactions between mRNA and tRNA [37][2]. Considering their ribosomal activity, compounds such as gentamicin, tobramycin, paromomycin, and geneticin (Figure 31) were also studied for their readthrough-inducing features.
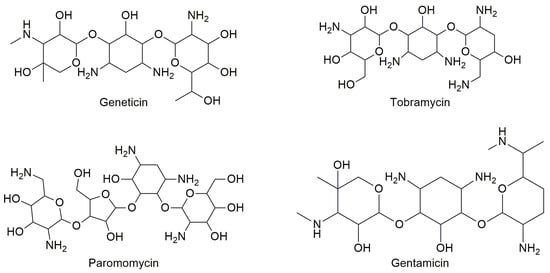
Figure 31.
Structures of some aminoglycoside antibiotics studied for readthrough ability.
Most studies have demonstrated that tobramycin has low efficiency as a PTC suppressor, while gentamicin is very efficient as a TRID although it presents an undesirable toxic profile because of its ototoxicity and nephrotoxicity [38][3]. Some studies have demonstrated that the activity of aminoglycoside TRIDs varies depending on the stop codon sequence (UGA, UAA, or UAG) [32,39,40][4][5][6]. Moreover, the negative side effects of this class of drugs as TRIDs are also due to the readthrough of correctly positioned stop codons, thus demonstrating a lack of selectivity towards the premature stop codon. Additionally, aminoglycoside antibiotics cannot be used for long-term therapy (as it would be required by their use as TRIDs) because of the risk of inducing bacterial resistance [37][2]. An interesting alternative to gentamicin is represented by ELX-02 (Figure 42), a synthetic aminoglycoside that binds eukaryotic ribosomes and has been recently claimed to induce the readthrough of PTCs without the toxicity of the antibiotics.
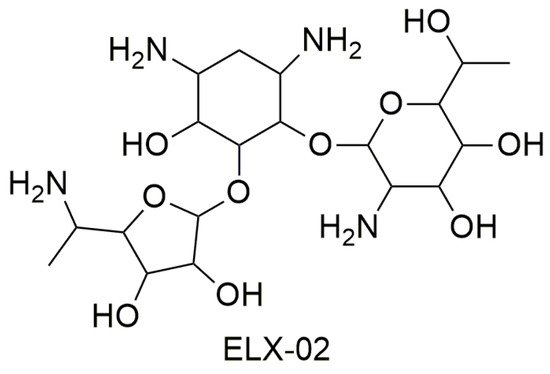
Figure 42.
Structure of ELX-02.
The first study showed that ELX-02 is capable both to restore the synthesis of the CFTR protein in the presence of nonsense mutations and to increase the levels of mRNA, which indicates that the drug is capable to interact with the NMD mechanism and/or to stabilize the transcript [37,41][2][7].
ELX-02 exercises its readthrough activity by stabilizing the “exo” conformation of two adenosine residues in rRNA. Because of this conformation, a near-cognate tRNA can bind the PTC, shifting aside eRF1. In this way, protein synthesis will continue beyond the PTC in order to generate a full-length protein. ELX-02 shows greater selectivity for cytoplasmic ribosomes than gentamicin, which results in increased readthrough activity and lower toxicity. In fact, aminoglycoside toxicity is determined by its affinity for mitochondrial ribosomes, and ELX-02’s affinity for this site is 100-fold lower than other aminoglycoside antibiotics. As for clinical studies, ELX-02 underwent randomized double-blind placebo-controlled trials. It was administered subcutaneously in doses of 0.3 to 7.5 mg/kg and intravenously in doses of 0.3 mg/kg. These studies confirmed that ELX-02 administered subcutaneously is widely bioavailable, well-tolerated, and does not present toxic effects, such as nephrotoxicity and ototoxicity when administered in doses within the therapeutic range [37][2].
2. Oxadiazoles
Oxadiazoles are aromatic heterocyclic rings containing one oxygen and two nitrogen atoms occupying different ring positions as in 1,2,3- [42][8], 1,2,4- [43[9][10],44], 1,2,5- [45][11], and 1,3,4-oxadiazoles [46,47][12][13] (Figure 53). Depending on the relative position of nitrogen and oxygen atoms, these classes of oxadiazoles possess different characteristics. For instance, 1,2,4-oxadiazoles have lower water solubility than 1,3,4-oxadiazoles because of the reduced hydrogen bond acceptor character of the nitrogen in 1,2,4-oxadiazole ring [43][9]. Moreover, they have been extensively studied because of their peculiar reactivity [43,48][9][14] and possible applications in material chemistry [49,50,51,52,53,54,55][15][16][17][18][19][20][21] or as bioactive compounds [44,56,57,58][10][22][23][24].
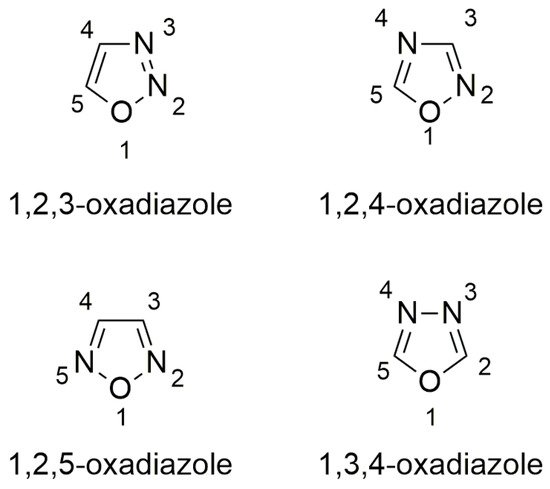
Figure 53.
Structures of oxadiazoles.
Of all the four possible classes of oxadiazoles, 1,2,4- and 1,3,4-oxadiazoles are the most studied for their pharmaceutical properties [59][25]. Their heterocyclic core is present in commercial drugs, such as the antitussive oxolamine (Perebron®) [60][26] and antiviral (Raltegravir®) [61][27], or potential anticancer drug candidates [62][28].
In 2007, a fluorinated 1,2,4-oxadiazole, specifically the 3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]-benzoic acid also known as Ataluren or PTC124 (Figure 64), was reported to promote the readthrough of nonsense mutations [33][29]. Ataluren is capable of promoting the readthrough of UGA, UAG, and UAA codons, but it shows the highest readthrough activity for the UGA codon [6,33][30][29]. Since Ataluren is structurally different from aminoglycosides, its discovery opened the way to a different class of drugs able to treat genetic disorders caused by nonsense mutations. Additionally, its activity profile within a 0.01–3 µM therapeutic range was better than that of gentamicin aminoglycoside, which was active as a TRID at much higher concentrations [6,33][30][29]. Furthermore, Ataluren is able to distinguish between normal and premature stop codons, thus inducing selective readthrough of PTCs without showing the typical toxicity of aminoglycosides. Indeed, several clinical trials have emphasized the safety and tolerability of Ataluren, even for long-term treatments [6,33,63][30][29][31].
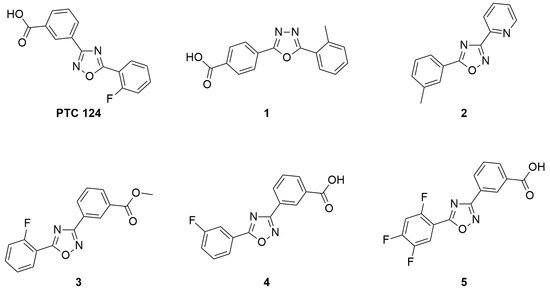
Figure 64.
Structure of Ataluren (also known as PTC124 or Translarna
®
) and its most active analogs.
Today, Ataluren is marketed under the trade name of Translarna® and has been approved as a therapy for ambulatory patients aged five years and older affected by Duchenne muscular dystrophy [64][32]. However, its use is not generally extensible to a wide range of cases since not all patients suffering from pathologies caused by nonsense mutations found beneficial effects, especially if Ataluren is administered in conjunction with other drugs [65][33]. For instance, in an Ataluren clinical trial performed on patients affected by cystic fibrosis, and taking tobramycin by inhalation, the competitive action between Ataluren and aminoglycoside antibiotics was highlighted [65][33]. This competition could be due to the fact that Ataluren also targets the ribosomal machinery as well as tobramycin, although the actual biomolecular target could be different for the two drugs. Indeed, Ataluren’s readthrough activity has been questioned [66[34][35][36],67,68], and its detailed mechanism of action is still debated, despite the fact that the functionality of rescued full-length proteins has been demonstrated [69,70][37][38] and the consensus is converging towards its interaction with mRNA [71][39]. Confirmation of Ataluren’s readthrough activity by orthogonal in vitro assays [72][40] prompted researchers to optimize this lead compound by designing Ataluren’s analogs that could be used for the treatment of diseases caused by nonsense mutations. The modification of Ataluren’s scaffold could involve either the oxadiazole heterocyclic core or the attached lateral moieties. In this context, and in the absence of a certain biomolecular target, computational optimization of Ataluren’s analogs could be achieved by ligand-based virtual screening. Thus, virtually selected sets of molecules containing either the 1,2,4- or the 1,3,4-oxadiazole core were synthesized and experimentally tested by FLuc cell-based assays to evaluate their readthrough ability [29,34,35,72][41][42][43][40]. In particular, Hela cells were transfected with pFLuc wild-type (pFLuc-wt) or pFLuc-opal (containing the UGA stop codon) plasmids. The expression of FLuc gene was then evaluated by measuring the luminescence of the produced luciferase in cells treated with the tested derivatives. However, Auld et al. [67][35] questioned the validity of FLuc cell-based assays as a method to evaluate the readthrough activity of Ataluren and its analogs, claiming that these compounds stabilized FLuc, protecting it from its degradation by trypsin. Therefore, an orthogonal assay was developed to confirm the readthrough activity of the new compounds that were the most active according to the Fluc assay (Figure 75A).
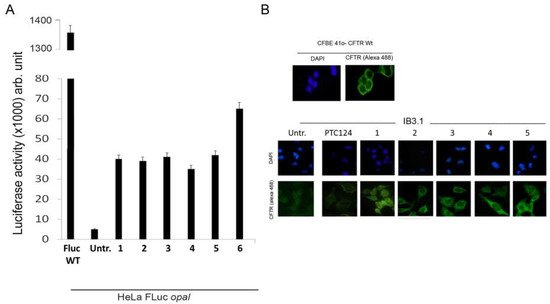
Figure 75. (A) Histogram of luciferase activity shown by HeLa cells after treatment with some of the most active compounds compared with untreated cells and with Fluc used as positive control; (B) Immunofluorescence of IB3.1 cells untreated (Untr; negative control) or treated with PTC124 (Ataluren; positive control) and compounds 1–5 respectively. Cystic fibrosis transmembrane regulator (CFTR) protein was revealed by a specific antibody targeting its first external loop (secondary antibody in green, Alexa-488). Nuclei (blue) were DAPI (4′,6-diamidino-2-phenylindole) stained.
To this aim, a reporter plasmid (H2B-GFP-opal), in which the cDNA of the H2B gene is fused to the cDNA of the green fluorescent protein (GFP) harboring a PTC at Trp58 [72][40], has been generated. Hela cells were then transfected with the normal (H2B-GFP) and the mutated (H2B-GFP-opal) plasmids and treated with selected compounds. The detection of H2BGFP by immunofluorescence demonstrated the synthesis of a full-length H2BGFP protein and hence the occurrence of readthrough. These results were confirmed also by western blotting, indicating the achievement of a full-length protein [29,34,35][41][42][43]. The most active compounds were also tested for nonsense suppression in the cystic fibrosis bronchial epithelial cell line IB3.1 derived from a CF patient. Also, in this case, fluorescence microscopy demonstrated the readthrough capacity of these compounds with the consequent restoring of full-length CFTR and its correct localization on the cell membrane (Figure 75B). Moreover, western blotting was used to quantify the CFTR level, and it confirmed the significant increase in protein level in cells treated with Ataluren analogs compared to untreated cells [29,34,35][41][42][43].
Once the readthrough activity of Ataluren-like compounds was also confirmed on the CF model, leading to a significant increase of CFTR expression (compared to untreated cells) and to its correct location on the cell membrane, the physiological functionality of the recovered protein needed to be assessed. For this purpose, Fisher rat thyroid (FRT) cells transfected with a plasmid vector harboring the mutated (G542X) CFTR were used to evaluate the expression and activity of the CFTR channel. Fluorescence microscopy images indicated the readthrough of the PTC and the correct position of CFTR at the cell membrane; western blotting allowed to quantify the level of CFTR protein. The functionality of the CFTR channel can be evaluated by two approaches: on one hand, FRT cells capable of differentiating in an epithelium can be used to measure the ion current of chloride ions crossing the epithelium in an Ussing flux chamber, thus giving a functionality response [73][44]. On the other hand, FRT cells expressing EYFP protein (an ectopically expressed mutant form of the yellow fluorescent protein) can be used for a quench-EYFP assay based on iodide-mediated fluorescence quenching to evaluate the halide ion flow across the cell membrane [74][45]. With both approaches an increase in the functionality of the chloride channel was observed when nonsense CFTR(G542X-opal) FRT cells were treated with compound one, demonstrating its activity as a TRID [35][43].
3. Miscellanea
Besides oxadiazoles and aminoglycosides, other small molecules have recently been tested as TRIDs to treat genetic disorders caused by nonsense mutations, and results concerning their activity are summarized below. Amlexanox (Figure 86), an antiallergic and anti-inflammatory agent that is administered orally or topically to treat asthma and aphthous ulcers [75,76[46][47][48],77], was discovered to inhibit NMD and induce PTC readthrough. Although this double-action has been previously described for geneticin as well as for other TRIDs, Amlexanox is less toxic and active at lower concentrations than those required for geneticin. Additionally, apparently, Amlexanox is somehow specific and does not interfere with naturally occurring NMD processes. In this way, the increased number of mRNAs surviving the NMD results in some recovery of a full-length protein [75][46].
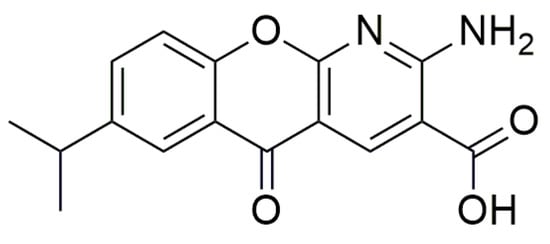
Figure 86.
Structure of Amlexanox.
Recently, the nucleoside analog Clitocine (Figure 97) was discovered to induce readthrough with a TRID activity two to three orders of magnitude higher than gentamicin and geneticin [78][49]. Furthermore, Clitocine has a greater selectivity towards PTCs than towards normal stop codons. Interestingly, to exert its readthrough activity, it is necessary that Clitocine is incorporated into mRNA after its conversion into Clitocine-5′-triphosphate. Friesen et al. [78][49] claim that, surprisingly, the incorporation of Clitocine in the mRNA sequence increased the readthrough of PTCs, and that readthrough activity was dose-dependent. Moreover, this nucleoside analog is able to induce the readthrough in tumors harboring a nonsense mutation onto the p53 gene, thus inhibiting the growth of tumors.
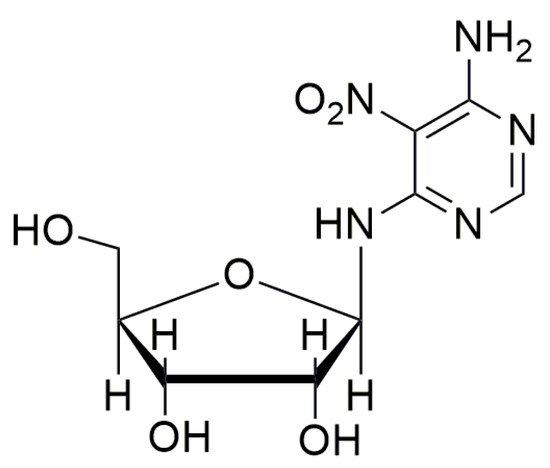
Figure 97.
Structure of Clitocine.
Other approaches to suppress the effect of nonsense mutations involve genetic engineering and include the administration of CRISPR-Cas9 (Crispr-ASsociated), a nuclease that cuts DNA in a sequence-specific manner guided by a complementary sequence of RNA (gRNA). This strategy, tested for the correction of the CFTR gene, did not lead to sufficient repair and introduced issues concerning the use of appropriate vectors for the administration of the complex [25][50]. On the other hand, a recent study reported the use of anticodon engineered transfer RNAs (ACE-tRNA) as a possible strategy to suppress PTCs and introduced a near-cognate amino acid in the growing peptide chain. Since ACE-tRNA showed limited interactions with normal stop codons, the use of these engineered biomolecules represents a promising approach for selective readthrough [79][51].
References
- Friesen, W.J.; Johnson, B.; Sierra, J.; Zhuo, J.; Vazirani, P.; Xue, X.; Tomizawa, Y.; Baiazitov, R.; Morrill, C.; Ren, H.; et al. The minor gentamicin complex component, X2, is a potent premature stop codon readthrough molecule with therapeutic potential. PLoS ONE 2018, 13, 1–18.
- Leubitz, A.; Frydman-Marom, A.; Sharpe, N.; van Duzer, J.; Campbell, K.C.M.; Vanhoutte, F. Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of ELX-02, a Potential Treatment for Genetic Disorders Caused by Nonsense Mutations, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 1–11.
- Bidou, L.; Bugaud, O.; Belakhov, V.; Baasov, T.; Namy, O. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol. 2017, 14, 378–388.
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055.
- Karijolich, J.; Yu, Y.T. Therapeutic suppression of premature termination codons: Mechanisms and clinical considerations (Review). Int. J. Mol. Med. 2014, 34, 355–362.
- Floquet, C.; Hatin, I.; Rousset, J.P.; Bidou, L. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals a major determinant of response to gentamicin. PLoS Genet. 2012, 8, e1002608.
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic Aminoglycosides Efficiently Suppress Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations and Are Enhanced by Ivacaftor. Am. J. Respir. Cell Mol. Biol. 2014, 50, 805–816.
- Fraser, W. 1, 2, 3-Oxadiazoles. In Comprehensive Heterocyclic Chemistry III, 1st ed.; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 5, p. 211.
- Pace, A.; Buscemi, S.; Palumbo Piccionello, A.; Pibiri, I. Recent Advances in the Chemistry of 1, 2, 4-Oxadiazoles. In Advances Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 116, pp. 85–136.
- Pace, A.; Pierro, P. The new era of 1, 2, 4-oxadiazoles. Org. Biomol. Chem. 2009, 7, 4337–4348.
- Paton, R.M. 1, 2, 5-Oxadiazoles. In Comprehensive Heterocyclic Chemistry II, 1st ed.; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 4, pp. 229–265.
- Salahuddin, A.; Mazumder, M.; Yar, M.S.; Mazumder, R.; Chakraborthy, G.S.; Ahsan, M.J.; Rahman, M.U. Updates on synthesis and biological activities of 1, 3, 4-oxadiazole: A review. Synth. Commun. 2017, 47, 1805–1847.
- Paun, A.; Hadade, N.D.; Paraschivescu, C.C.; Matache, M. 1, 3, 4-Oxadiazoles as luminescent materials for organic light emitting diodes via cross-coupling reactions. J. Mater. Chem. C 2016, 4, 8596–8610.
- Palumbo Piccionello, A.; Pace, A.; Buscemi, S. Rearrangements of 1, 2, 4-Oxadiazole: “One Ring to Rule Them All”. Chem. Heterocycl. Compd. 2017, 53, 936–947.
- Palumbo, F.S.; Di Stefano, M.; Palumbo Piccionello, A.; Fiorica, C.; Pitarresi, G.; Pibiri, I.; Buscemi, S.; Giammona, G. Perfluorocarbon functionalized hyaluronic acid derivatives as oxygenating systems for cell culture. RSC Adv. 2014, 4, 22894–22901.
- Pibiri, I.; Pace, A.; Buscemi, S.; Causin, V.; Rastrelli, F.; Saielli, G. Oxadiazolyl-pyridines and perfluoroalkyl-carboxylic acids as building blocks for protic ionic liquids: Crossing the thin line between ionic and hydrogen bonded materials. Phys. Chem. Chem. Phys. 2012, 14, 14306–14314.
- Palumbo Piccionello, A.; Calabrese, A.; Pibiri, I.; Giacalone, V.; Pace, A.; Buscemi, S. Synthesis of Fluorinated Bent-Core Mesogens (BCMs) Containing the 1, 2, 4-Oxadiazole Ring. J. Heterocycl. Chem. 2016, 53, 1935–1940.
- Palumbo Piccionello, A.; Guarcello, A.; Calabrese, A.; Pibiri, I.; Pace, A.; Buscemi, S. Synthesis of fluorinated oxadiazoles with gelation and oxygen storage ability. Org. Biomol. Chem. 2012, 10, 3044–3052.
- Pibiri, I.; Beneduci, A.; Carraro, M.; Causin, V.; Casella, G.; Corrente, G.A.; Chidichimo, G.; Pace, A.; Riccobono, A.; Saielli, G. Mesomorphic and electrooptical properties of viologens based on non-symmetric alkyl/polyfluoroalkyl functionalization and on an oxadiazolyl-extended bent core. J. Mater. Chem. C 2019.
- Huang, X.C.; Wang, Z.J.; Guo, T.; Qin, M.N.; Liu, M.; Qiu, S.J. Review on Energetic Compounds Based on 1, 2, 4-Oxadiazoles. Hanneng Cailiao/Chin. J. Energetic 2017, 25, 603–611.
- Fouad, F.S.; Ness, T.; Wang, K.; Ruth, C.E.; Britton, S.; Twieg, R.J. Biphenylyl-1, 2, 4-oxadiazole based liquid crystals–synthesis, mesomorphism, effect of lateral monofluorination. Liq. Cryst. 2019.
- Palumbo Piccionello, A.; Musumeci, R.; Cocuzza, C.; Fortuna, C.G.; Guarcello, A.; Pierro, P.; Pace, A. Synthesis and preliminary antibacterial evaluation of Linezolid-like 1, 2, 4-oxadiazole derivatives. Eur. J. Med. Chem. 2012, 50, 441–448.
- Tarasenko, M.; Duderin, N.; Sharonova, T.; Baykov, S.; Shetnev, A.; Smirnov, A.V. Room-temperature synthesis of pharmaceutically important carboxylic acids bearing the 1, 2, 4-oxadiazole moiety. Tetrahedron Lett. 2017, 58, 3672–3677.
- Tarasenko, M.; Sidneva, V.; Belova, A.; Romanycheva, A.; Sharonova, T.; Baykov, S.; Shetnev, A.; Kofanov, E.; Kuznetsov, M.A. An efficient synthesis and antimicrobial evaluation of 5-alkenyl-and 5-styryl-1, 2, 4-oxadiazoles. Arkivoc 2018, 458–470.
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in Medicinal Chemistry. J. Med. Chem. 2012, 55, 1817–1830.
- Rottini, E.; Marri, G.; Calandra, P. The recent introduction in therapy of a new antitussive drug: Oxolamine. Minerva Med. 1961, 52, 3758–3760.
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Paz, O.G.; Hazuda, D.J.; et al. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855.
- James, N.D.; Growcott, J.W. Zibotentan. Drugs Fut. 2009, 34, 624.
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, T.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–93.
- Peltz, S.W.; Morsy, M.; Welch, E.M.; Jacobson, A. Ataluren as an Agent for Therapeutic Nonsense Suppression. Annu. Rev. Med. 2013, 64, 407–425.
- Hirawat, S.; Welch, E.M.; Elfring, G.L.; Northcutt, V.J.; Paushkin, S.; Hwang, S.; Leonard, E.M.; Almstead, N.G.; Ju, W.; Peltz, S.W.; et al. Safety, Tolerability, and Pharmacokinetics of PTC124, a Nonaminoglycoside Nonsense Mutation Suppressor, Following Single- and Multiple-Dose Administration to Healthy Male and Female Adult Volunteers. J. Clin. Pharmacol. 2007, 47, 430–444.
- Haas, M.; Vlcek, V.; Balabanov, P.; Salmonson, T.; Bakchine, S.; Markey, G.; Weise, M.; Schlosser-Weber, G.; Brohmann, H.; Yerro, C.P.; et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul. Disord. 2015, 25, 5–13.
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547.
- Siddiqui, N.; Sonenberg, N. Proposing a mechanism of action for ataluren. Proc. Natl. Acad. Sci. USA 2016, 113, 12353–12355.
- Auld, D.S.; Thorne, N.; Maguire, W.F.; Inglese, J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. USA 2009, 106, 3585–3590.
- Auld, D.S.; Lovell, S.; Thorne, N.; Lea, W.A.; Maloney, D.J.; Shen, M.; Rai, G.; Battaile, K.P.; Thomas, C.J.; Simeonov, A.; et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc. Natl. Acad. Sci. USA 2010, 107, 4878–4883.
- Roy, B.; Leszyk, J.B.; Mangus, D.A.; Jacobson, A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. USA 2015, 112, 3038–3043.
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA 2016, 113, 12508–12513.
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med. Chem. Lett. 2019, 10, 522–527.
- Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; Palumbo Piccionello, A.; Pibiri, I. Toward a Rationale for the PTC124 (Ataluren) Promoted Readthrough of Premature Stop Codons: A Computational Approach and GFP-Reporter Cell-Based Assay. Mol. Pharm. 2014, 11, 653–664.
- Pibiri, I.; Lentini, L.; Melfi, R.; Gallucci, G.; Pace, A.; Spinello, A.; Barone, G.; Di Leonardo, A. Enhancement of premature stop codon readthrough in the CFTR gene by Ataluren (PTC124) derivatives. Eur. J. Med. Chem. 2015, 101, 236–244.
- Pibiri, I.; Lentini, L.; Tutone, M.; Melfi, R.; Pace, A.; Di Leonardo, A. Exploring the readthrough of nonsense mutations by non-acidic Ataluren analogues selected by ligand-based virtual screening. Eur. J. Med. Chem. 2016, 122, 429–435.
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142.
- Gitter, A.H.; Schulzke, J.D.; Sorgenfrei, D.; Fromm, M. Ussing chamber for high-frequency transmural impedance analysis of epithelial tissues. J. Biochem. Biophys. Methods 1997, 35, 81–88.
- Baumann, C.T.; Lim, C.S.; Hager, G.L. Simultaneous Visualization of the Yellow and Green Forms of the Green Fluorescent Protein in Living Cells. J. Histochem. Cytochem. 1998, 46, 1073–1076.
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Déprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 1–14.
- Saijo, T.; Kuriki, H.; Ashida, Y.; Makino, H.; Maki, Y. Mechanism of the action of Amoxanox (AA-673) an Orally Active Antiallergic Agent. Int. Archs Allergy Appl. Immun. 1985, 78, 43–50.
- Meng, W.; Dong, Y.; Liu, J.; Wang, Z.; Zhong, X.; Chen, R.; Zhou, H.; Lin, M.; Jiang, L.; Gao, F.; et al. A clinical evaluation of amlexanox oral adhesive pellicles in the treatment of recurrent aphthous stomatitis and comparison with amlexanox oral tablets: A randomized, placebo controlled, blinded, multicenter clinical trial. Trials 2009, 10.
- Friesen, W.J.; Trotta, C.R.; Tomizawa, Y.; Zhuoa, J.; Johnson, B.; Sierra, J.; Roy, B.; Weetall, M.; Hedricka, J.; Sheedy, J.; et al. The nucleoside analog clitocine is a potent and efficacious readthrough agent. RNA 2017, 23, 567–577.
- Colemeadow, J.; Joyce, H.; Turcanu, V. Precise treatment of cystic fibrosis–current treatments and perspectives for using CRISPR. Expert Rev. Precis. Med. Drug Dev. 2016, 1, 169–180.
- Lueck, J.D.; Yoon, J.S.; Perales-Puchalt, A.; Mackey, A.L.; Infield, D.T.; Behlke, M.A.; Pope, M.R.; Weiner, D.B.; Skach, W.R.; McCray, P.B., Jr.; et al. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 2019, 10.
More