Despite recent improvements in diagnostic ability and treatment strategies, advanced gastric cancer (GC) has a high frequency of recurrence and metastasis, with poor prognosis. To improve the treatment results of GC, the search for new treatment targets from proteins related to epithelial–mesenchymal transition (EMT) and cell–cell adhesion is currently being conducted. EMT plays an important role in cancer metastasis and is initiated by the loss of cell–cell adhesion, such as tight junctions (TJs), adherens junctions, desmosomes, and gap junctions. Among these, claudins (CLDNs) are highly expressed in some cancers, including GC. Abnormal expression of CLDN1, CLDN2, CLDN3, CLDN4, CLDN6, CLDN7, CLDN10, CLDN11, CLDN14, CLDN17, CLDN18, and CLDN23 have been reported. Among these, CLDN18 is of particular interest. In The Cancer Genome Atlas, GC was classified into four new molecular subtypes, and CLDN18–ARHGAP fusion was observed in the genomically stable type. An anti-CLDN18.2 antibody drug was recently developed as a therapeutic drug for GC, and the results of clinical trials are highly predictable. Thus, CLDNs are highly expressed in GC as TJs and are expected targets for new antibody drugs.
1. Introduction
Gastric cancer (GC) is one of the most common cancers and the third leading cause of cancer-related deaths worldwide, with a high incidence in East Asia
[1]. Despite advances in diagnostic equipment and treatment strategies, GC has a poor prognosis, with recurrence and metastasis. Common metastatic sites for GC include the peritoneum, lymph nodes, liver, lungs, and bones
[2][3][2,3].
The first step in cancer metastasis is local infiltration into the surrounding tumor-related stroma and normal tissue. In this metastatic process, epithelial–mesenchymal transition (EMT) is an important step for cancer cells to acquire a metastatic phenotype in the state of mesenchymal cells
[4]. The first stage of EMT is the breakdown of contact between epithelial cells, such as tight junctions (TJs), adherens junctions, desmosomes, and gap junctions, eventually resulting in the loss of cell polarity, cytoskeletal reorganization, and cells. Among these contacts, TJs comprise endogenous membrane proteins, such as claudin (CLDN) and occludin, and cytoplasmic proteins zonula occludens 1 (ZO1), ZO2, and ZO3, which are transmembrane proteins. They link the actin cytoskeleton and signaling proteins
[4][5][6][4,5,6]. In general, CLDNs play an essential role in scaffolding for cell–cell adhesion and migration. The expression of CLDN has been reported in several types of cancer, including GC, and has recently attracted attention as a new therapeutic target
[7]. In 2014, The Cancer Genome Atlas (TCGA) classified GC into four subtypes: Epstein–Barr virus (EBV)-positive, microsatellite instability, genomic stability (GS), and chromosomal instability. Among these, it was reported that the characteristics of GS are rich in mutations in the fusion of
CLDN18–
ARHGAP, in addition to the diffuse histological type and
RHOA [8]. Furthermore, the efficacy of anti-CLDN18 antibody drugs in patients with GC has been reported, and clinical trials are currently underway
[9][10][9,10].
2. General Information of CLDNs
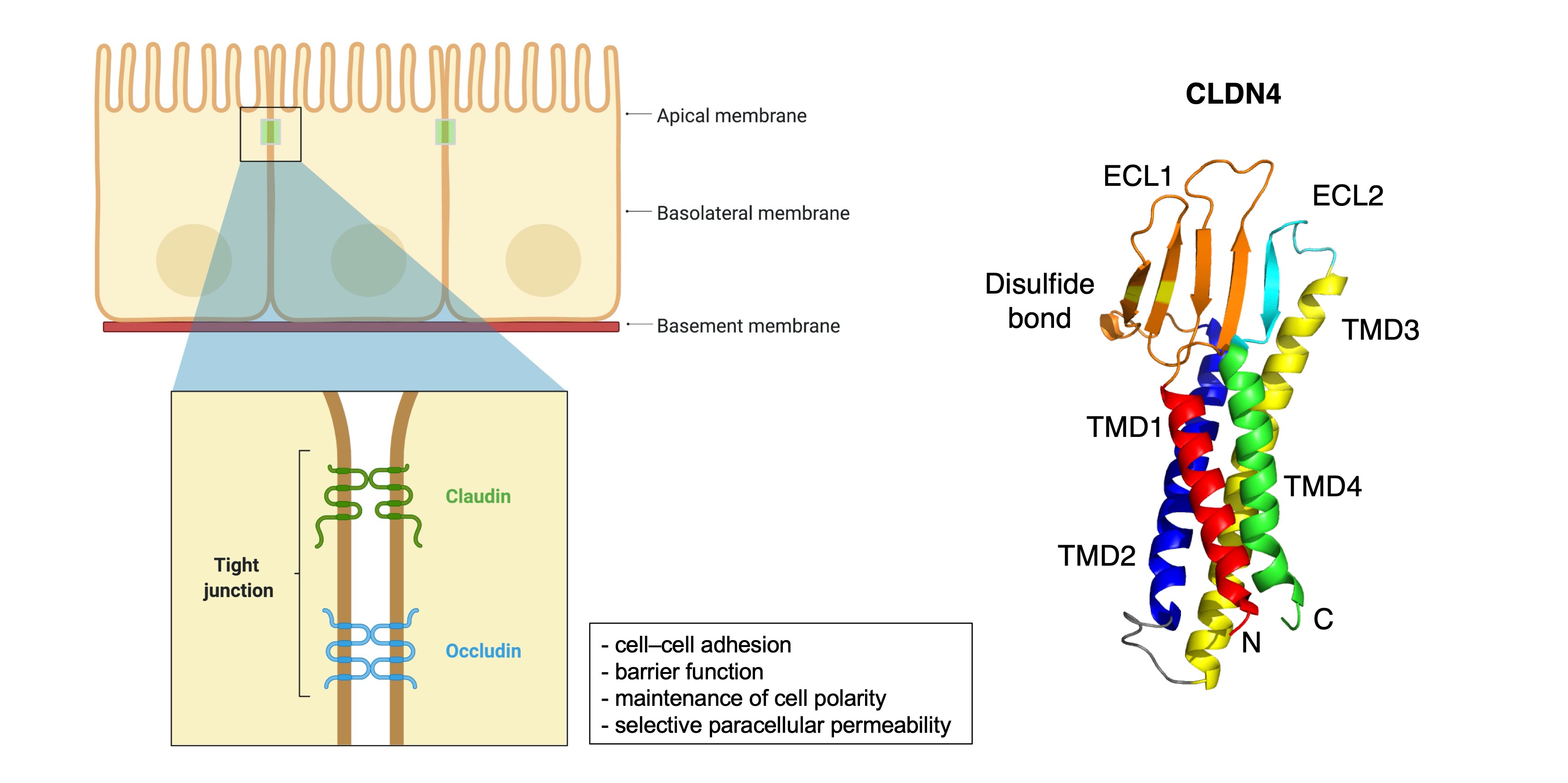
CLDNs are a family of at least 27 transmembrane proteins
[11][12][11,12] and were first reported by Tsukita et al.
[13]. CLDNs are classified into classic and non-classic types according to their difference in sequence
[11][14][11,14]. Classic types include CLDNs1–10, CLDN14, CLDN15, CLDN17, CLDN19, and non-classic types include CLDNs11–13, CLDN16, CLDN18, and CLDNs20–24
[14]. CLDNs are 20–34 kDa in size
[14] and are composed of the N-terminal region of the cytoplasm, two extracellular loops, four transmembrane domains, and the C-terminal tail of the cytoplasm
[7][14][7,14] (
Figure 1). The large extracellular loops of CLDNs have charged amino acids and form charge-selective channels that regulate the ion selectivity of neighboring cells
[15]. The two cysteines of CLDNs may form internal disulfide bonds to stabilize protein conformation
[16], and the shorter second extracellular loop is folded with a helix-turn-helix motif. It may be involved in dimer formation between CLDNs on opposite cell membranes through hydrophobic interactions between conserved aromatic residues
[17]. The C-terminus of the CLDN protein exhibits diversity in sequence and length. Most CLDN family members provide scaffolding for migration, cell adhesion, matrix remodeling, and proliferation. It forms a C-terminal PDZ-binding motif that interacts with ZO1, ZO2, ZO3, and multi-PDZ domain protein 1 (MUPP1)
[18][19][20][18,19,20]. This region also contains amino acid residues associated with post-translational modifications such as serine–threonine phosphorylation, tyrosine phosphorylation, SUMOylation, and palmitoylation, all of which affect the localization and function of CLDNs
[21][22][23][24][25][21,22,23,24,25].
Figure 1. Schematic of the claudin protein located in tight junction and the 3D structures of CLDN4 (PDB: 7KP4). Adapted from “Cell Junction Types”, by BioRender.com (2021). Retrieved from
https://app.biorender.com/biorender-templates (accessed on 30 September 2021).
Furthermore, the phosphorylation of the C-terminal tail of CLDNs interacts with several major kinases. CLDN3 is phosphorylated by Protein Kinase A, which induces disruption of the TJs
[26]. CLDN5 has been reported to increase the permeability of Palauan endothelium through phosphorylation via protein kinases Cα and ζ
[27]. CLDNs1–4 are phosphorylated by the threonine/serine kinase WNK4
[28], and CLDN4 is phosphorylated at the PDZ-binding C-terminus by the receptor tyrosine kinase EphA2, which reduces the binding to ZO1 and is intercellular and promotes contact withdrawal
[29]. The phosphorylation of CLDNs as described above may downregulate TJ intensity
[26][30][26,30].
From a structural and functional view, CLDNs generally localize to the apical region of the cell membrane and form a TJ complex for cell–cell adhesion, maintenance of cell polarity, and selective paracellular permeability, and play an important role in barrier function
[6][31][32][33][34][35][6,31,32,33,34,35]. As loss of the cell–cell adhesion complex is associated with increased EMT in cancer, phosphorylation and reduction of CLDNs may promote metastasis and infiltration. Alternatively, overexpression of CLDNs has also been reported to increase aberrant localization and function in gastric, lung, prostate, ovarian, colorectal and breast cancers, promoting metastasis and progression
[7].
Thus, CLDNs are generally highly expressed in cancer tissues depending on the type of cancer and the type of CLDN, although their reduction or loss of function due to phosphorylation promotes EMT and cancer metastasis and infiltration.
3. CLDN Expression in GC
The expression of several CLDNs has been described in GC, including their functions, molecules involved, and clinical significances (
Table 1). The following section summarizes new insights and deeper considerations for each CLDN’s expression in GC.
3.1. CLDN18
The CLDN18 protein is the most studied among the CLDNs, since it is specifically expressed in stomach and GC tissue, compared to other tissues or cancers, which makes it a potential therapeutic target. CLDN18 was first identified in 2001 as a downstream target gene of the T/EBP/NKX2.1 homeodomain transcription factor
[36]. The human
CLDN18 gene has two splice variants, which encode two protein isoforms, CLDN18 splice variant 1 (CLDN18.1) and CLDN18 splice variant 2 (CLDN18.2)
[37]. CLDN18.1 is specifically expressed in normal and cancerous lung tissues, and CLDN18.2 is expressed in normal gastric tissues and in tissues of gastric, pancreatic, esophageal, and lung cancers
[37]. In the normal gastric mucosa, CLDN18 is found on the surface of the lobular epithelium, immature cells, and glandular epithelium. In intestinal metaplasia, CLDN18 is absent in metaplastic epithelium
[38].
Table 1. Claudin’s functions, signaling molecules involved, and clinical significances in GC.
| Type of CLDN |
Main Functions, Signaling Molecules Involved, and Clinical Significances in GC |
References |
| CLDN1 |
- correlation with tumor infiltration and metastasis
- Akt, Src, and NF-κB signaling pathway
- poor prognostic factor |
[39][40][39,40]
[41][42][41,42]
[40][43][40,43] |
| CLDN2 |
- CDX2-dependent targeting relationship with CagA produced from H. pylori |
[44] |
| CLDN3 |
- correlation with lymph node metastasis
- important immunosuppressive regulator |
[45]
[46] |
| CLDN4 |
- correlation with lymph node metastasis
- promoting EMT and infiltration of MMP-2 and MMP-9 |
[47]
[48][49][50][48,49,50] |
| CLDN6 |
- cell proliferation and migration/infiltration with YAP1
- high expression was positively correlated with decreased OS |
[51]
[51][52][51,52] |
| CLDN7 |
- proliferation in a CagA-and β-catenin-dependent manner
- poor prognostic factor |
[53]
[54] |
| CLDN10 |
- association with metastasis and proliferation |
[55][56][55,56] |
| CLDN11 |
- correlation with H. pylori infection and Borrmann classification, not with lymph node metastasis and TNM stage |
[57] |
| CLDN14 |
- correlation with lymph node metastasis |
[58] |
| CLDN17 |
- correlation with lymph node metastasis |
[58] |
| CLDN18 |
- correlation with metastasis (lymph node, peritoneal, bone, and liver metastasis)
- poor prognostic factor
- Wnt, β-catenin, CD44, EFNB/ EPHB receptor signals, and HIPPO signals
- CLDN18-ARHGAP fusion in genomically stable type
- therapeutic target of Claudiximab (IMAB362, Zolbetuximab) |
[59][60][59,60]
[61][62][63][64][65][66][67][68][61,62,63,64,65,66,67,68]
[54][69][54[70],69,70]
[8]
[10][71][72][73][10,71,72,73] |
| CLDN23 |
- poor prognostic factor |
[57] |
Downregulation of CLDN18 has been reported to occur during intestinal metaplasia
[74][75][74,75] in the Correa cascade, a multistep and multifactorial process of gastric carcinogenesis
[69][70][69,70]. Notably, the expression of CLDN18 in GCs has different biological functions depending on whether it is up- or downregulated; CLDN18 is also highly expressed in normal gastric tissues, and downregulation of this expression was detected in 57.5% of GCs, especially in 73.7% of intestinal phenotypes
[69]. Similarly, CLDN18 was downregulated in GC compared to the normal mucosa of the surrounding stomach and intestinal metaplasia
[54][76][54,76].
In the relationship between CLDN18 expression and clinicopathological factors in GC, CLDN18 expression was significantly lower in patients with peritoneal metastasis (PM) than those without PM (
p = 0.01). Meanwhile, CLDN18 expression was significantly higher in patients with bone metastasis than in those without bone metastasis (
p = 0.01)
[59]. Another IHC study showed that CLDN18 expression correlated with lymph node metastasis (
p = 0.04), high stage disease (III, IV) (
p = 0.019), and lower incidence of liver metastases (
p = 0.009)
[60]. In addition, a vital relationship existed between the decreased expression of CLDN18 and perineural invasion
[54].
In tissues of early-stage GC removed via endoscopic mucosal resection or endoscopic submucosal resection, the Ki-67 labeling index at the invasive front inversely correlated with the CLDN18 expression level, suggesting that a decrease in CLDN18 expression promotes cancer invasion in early-stage GC
[76]. Furthermore, concerning the relationship between CLDN18 expression and survival, patients without CLDN18 expression reported to have shorter overall survival (OS) than those with CLDN18 expression
[54][69][70][54,69,70].
To elucidate the biological significance and carcinogenic mechanism of reduced CLDN18 expression in GC, studies were conducted on GC cell lines in which CLDN18 was knocked down by siRNA and knocked out in mice
[76][77][76,77]. GC cells knocked down by CLDN18 promoted gastric cancer cell proliferation and infiltration compared to the controls
[76]. In CLDN18 knockout mice, CLDN18 was reported to be localized to TJs at the base rather than at the apex of gastric epithelial cells
[77]. Furthermore, in CLDN18 knockout mice, gastric mucosal atrophy and convulsive polypeptide expression alteration (SPEM) occur after paracellular H
+ ion leakage and parietal cell death, and SPEM was suggested as the origin of cancer stem cells
[78].
However, it has also been reported that CLDN18 knockout mice do not progress to cancer while maintaining atrophic gastritis and SPEM status
[79]. In other mouse models, CLDN18 deficiency occurred in the gastric mucosa of
Helicobacter pylori-infected mice
[77]. In contrast, the inactivation of CLDN18 in mice not infected by
H.
pylori promotes the growth of intraepithelial neoplasms that develop into polypoid tumors, β-catenin
[61][62][61,62], CD44
[63], and EFNB/ EPHB receptor signals
[64][65][64,65]. HIPPO signals
[66][67][66,67] and other vital signaling pathways promote cell proliferation, cancer stem cell development, and tumorigenesis. In GC CLDN18 knockout mice, chronic active gastritis developed at middle age (>40 weeks), and the expression of CCL28, a chemokine with lymphocyte chemo-ventilation activity, was observed. At old age (60 weeks or older), 20–30% of these mice develop gastric tumors, and CXCL5 is expressed. These are multifunctional cytokines with neutrophil-attracting, angiogenic, and EMT-inducing effects. During this process, SPEM cells developed, and the expression of CD44-variants, TLR2, and CXCL5 increased. Some features of gastric tumorigenesis in CLDN18 knockdown mice resemble human carcinogenesis associated with
H.
pylori infection. Wnt1 overexpression in transgenic CLDN18 knockout mice promotes gastric tumorigenesis. This indicates that when gastritis is induced by CLDN18 deficiency, Wnt-dependent gastric tumorigenesis may be triggered
[68].
While CLDN18 downregulation is crucial for GC development, proliferation, infiltration, and EMT, positive expression or upregulation of CLDN18 also plays a biologically important role in GC. In immunohistochemistry (IHC) studies, CLDN18 expression was found in 42.2% of GCs and correlated with the mucin phenotype, EBV status, integrin αvβ5, EpCAM extracellular domain EpEX, and lysozymes
[80]. Bioinformatics analysis of
CLDN18 expression in GC patients using multiple public databases revealed that
CLDN18 expression was higher in the EBV-positive group than in other groups
[81]. Tissue microarray analysis showed high membranous CLDN18 expression in 29.4% of primary cases and 34.1% of metastatic cases. Positive expression of membrane-type CLDN18 was significantly associated with the Lauren diffuse type (
p = 0.009) and EBV-related cancers (
p < 0.001)
[82]. In other IHC studies, CLDN18 was frequently expressed at the primary site, GC. In the metastatic cohort, 88% of GCs were positive for CLDN18
[83].
The TCGA Research Network
[8] revealed that the
CLDN18–
ARHGAP26/6 translocation was enriched in genomically stable tumors, which was also confirmed in diffuse GC
[84]. In the East Asian GC population,
CLDN18–
ARHGAP26/6 was detected in 3% of all cases
[85]. In other aspects, this unique fusion gene is associated with younger patients and invasive diseases, including lymph node and distant metastases
[86][87][86,87]. Histologically, signet ring cell carcinoma (SRCC), which is based on microscopic characteristics according to the classification by the World Health Organization, belongs to the diffuse type
[88]. The whole-genome sequence of 32 SRCC samples from the eastern Chinese population confirmed that the frequency of
CLDN18–
ARHGAP26/6 fusion was 25%. In the validation cohort, patients with
CLDN18–
ARHGAP26/6 fusion had poorer survival than those without fusion. Moreover, they did not benefit from oxaliplatin/fluoropyrimidine-based chemotherapy
[89]. This is consistent with the chemical drug resistance acquired in GC cells overexpressing
CLDN18–
ARHGAP26.
The subtype of gastric intestinal adenocarcinoma with anastomotic glands has been reported to be frequently associated with poorly differentiated adenocarcinoma components
[90][91][90,91]. In recent years,
RHOA mutations and
CLDN18–
ARHGAP fusions are typically present in adenocarcinomas with anastomotic glands via next-generation sequencer and reverse transcription-PCR
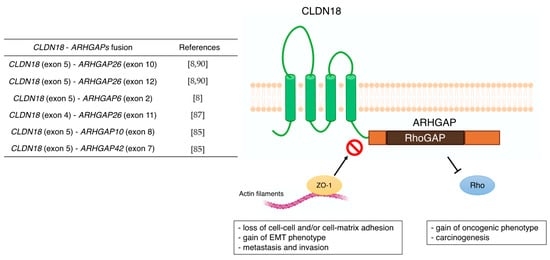
[92]. This characteristic chimeric protein CLDN18–ARHGAP in GC may also be associated with carcinogenesis through functional changes, such as the disappearance of CLDN18 and the acquisition of ARHGAP function (
Figure 2). Furthermore, the carboxyl-terminal PDZ-binding motif of CLDN18 interacts with actin-regulating proteins such as RhoA, cadherin, and integrins
[93]. Therefore, loss of the carboxyl-terminal domain of CLDN upon fusion with ARHGAP may prevent actin-regulated proteins from binding to the junctional complex and loosen cell–cell and/or cell–matrix binding. This aberrant ectopic activity of ARHGAP may be involved in carcinogenesis, just as the constitutive inactivation of RhoA leads to the acquisition of a carcinogenic phenotype
[94][95][94,95]. Alternatively, there are reports that mutations in
RHOA are gain-of-function
[84][96][84,96], and further studies are needed concerning the mechanism and effects of this unique fusion of
CLDN18–
ARHGAP in GC. Several cancer cell lines stably expressing
CLDN18–
ARHGAP26 showed a dramatic loss of the epithelial phenotype and long protrusions showing EMT. Fusion-positive cell lines showed loss of barrier properties, reduced cell–cell and cell–extracellular matrix adhesion, delayed wound healing, RhoA inhibition, and acquisition of invasiveness. Thus,
CLDN18–
ARHGAP26 contributes to epithelial degradation, possibly causing H
+ leakage in the stomach, inducing inflammation, and promoting infiltration
[85]. In addition to the
CLDN18 (exon5)-
ARHGAP26 (exon12 or exon10) or
ARHGAP6 (exon2), fusion patterns reported in TCGA cohort
[8] and other reports
[87][92][87,92] have shown
CLDN18 (exon5)–
ARHGAP26 (exon12 or exon10) or
ARHGAP6 (exon2). There are rare cases of
CLDN18 (exon4)–
ARHGAP26 (exon11)
[89],
CLDN18 (exon5)–
ARHGAP10 (exon8), and
CLDN18 (exon5)–
ARHGAP42 (exon7)
[87]. Notably, all these
CLDN18–
ARHGAP fusions share a common RhoGAP domain after gene translocation and are thought to promote carcinogenesis and cancer progression by inactivating Rho.
Figure 2. Previous reports of CLDN18-ARHGAPs and schematic of CLDN18-ARHGAP protein. Created with BioRender.com.
CLDN 18.2 has been reported as a target for therapeutic antibodies
[36][37][97][98][99][36,37,97,98,99]. In normal gastric tissue, CLDN18.2 is contained in TJ supramolecular complexes of gastric mucosal cells, and the epitope of CLDN18.2 has little access to intravenous antibodies
[36][37][100][36,37,100]. However, the loss of cell polarity associated with malignancy exposes the epitope of CLDN18.2, making it accessible to bound antibodies, and CLDN18.2, which is maintained in GC and gastric metastases
[37][54][101][37,54,101]. Claudiximab (IMAB362, Zolbetuximab) is a novel chimeric IgG1 antibody that is highly specific for CLDN18.2. Claudiximab is derived from a mouse monoclonal antibody and is chimeric with the human IgG1 constant region for clinical application (
Table 2). This novel antibody binds to CLDN18.2 on the surface of cancer cells and stimulates cells and immune effectors that promote antibody-dependent cellular cytotoxicity and complement-dependent cellular cytotoxicity
[71]. Moreover, it can induce apoptosis and suppress cell proliferation. Combination therapy with Claudiximab and chemotherapy may promote T-cell infiltration and induce inflammatory cytokines
[32].
Table 2. Clinical trials associated with IMAB362.
| Study Name |
NCT Number |
Phase |
Number of Participants |
Design |
Response Rate |
OS |
PFS |
Adverse Effects |
| - |
NCT00909025 |
I |
15 |
Single-dose escalation study evaluating safety and tolerability |
- |
- |
- |
Vomiting |
| PILOT |
NCT01671774 |
I |
32 |
Multiple dose study of IMAB362 with immunomodulation (Zoledronic acid and IL-2) |
11 patients had disease control |
40 weeks |
12.7 weeks |
Nausea and vomiting |
| MONO |
NCT01197885 |
IIa |
54 |
Multiple dose study of IMAB362 as monotherapy |
Clinical benefit rate: 23% |
- |
- |
Nausea, vomiting, and fatigue |
| FAST |
NCT01630083 |
IIb |
246 |
Randomized EOX vs. IMAB362 + EOX, extended with high-dose IMAB362 + EOX |
Objective response rate: 25 vs. 39% |
8.3 vs. 13.0 months |
5.3 vs. 7.5 months |
Neutropenia, anemia, weight loss, and vomiting |
| GLOW |
NCT03653507 |
III |
500 (estimated) |
Double-blinded, Randomized, IMAB362 plus CAPOX compared with placebo plus CAPOX as first-line treatment of subjects with CLDN 18.2-positive, HER2-negative locally advanced unresectable or metastatic gastric or gastroesophageal junction adenocarcinoma |
- |
- |
- |
- |