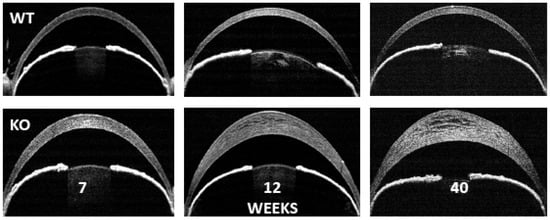
The apparent enhancement of H
+ flux when endothelial cells are perfused with NH
4Cl
[12] led to a series of patch-clamp studies to try to determine the nature of this enhanced flux. Using SLC4A11 transfected PS120 fibroblasts, Zhang et al.
[33][53] demonstrated inward H
+ currents in response to 10 mM NH
4Cl that increased from pH 6.5 to 7.5 to 8.5. Further analysis indicated that the currents were increasing with increasing [NH
3], but not [NH
4+] or [H
+]. If the [NH
3] was held constant at each pH, the inward current was approximately the same, suggesting that SLC4A11 is activated by increasing [NH
3] and not pH. These inward H
+ currents were not Na
+-dependent and were insensitive to EIPA
[33][53]. Kao et al.
[3] also showed NH
4Cl-sensitive inward currents in SLC4A11-transfected HEK cells that were, in contrast, mildly inhibited by EIPA. Using two electrode voltage clamps of oocytes transfected with mouse Slc4a11, Loganathan et al.
[11] observed similar inward H
+ currents stimulated by NH
4Cl, but concluded that SLC4A11 transports NH
3 rather than NH
3-H
+. Myers et al.
[34][54], also using transfected oocytes, showed enhanced Na
+-independent H
+ conduction, but concluded that SLC4A11 was activated by alkaline pH. Moreover, the H
+ conductance of SLC4A11 was found to be steepest at pH 8.5 and it was found that mutants can shift the optimum pKa
[35][55]. In summary, from these studies, it appears that SLC4A11 provides H
+ conductance that can be either Na
+ dependent or independent, is sensitive to NH
4Cl, and is stimulated by alkaline pH. Zhang et al.
[33][53] proposed that SLC4A11 is an NH
3/H
+ cotransporter. However, this does not rule out SLC4A11 being an NH
3 activated H
+ transporter, as demonstrating NH
3 net fluxes is difficult. The mechanism of an NH
3 activation is unknown, but could possibly be caused by changing the charge of pore amino acid residues that could also be accomplished by alkaline pH. A more recent study by Kao et al.
[9], using SLC4A11 transfected HEK cells, demonstrated H
+(OH
−) conductive transport stimulated by alkaline pH. Ammonia-stimulated currents were also increased in alkaline pH. The shift in the reversal potential from NH
3 suggested NH
3-H
+ cotransport was competing with H
+(OH
−) and the data, fitting a theoretical model of NH
3-H
+ and H
+(OH
−) interacting competitively within the transporter.
One of the features of the SLC4 family of transporters is their interaction with stilbene derivatives. SLC4A11 binding to stilbenes has been useful in membrane fractionation studies
[37][40]. Whereas stilbenes generally inhibit transport activity, Kao et al.
[16] showed an enhancement of H
+ flux by the stilbenes, H
2DIDS, SITS, and DIDS, which was also effective in increasing H
+ transport of the R109H mutant. Zhang et al.
[33][53] found that DIDS had no effect on NH
4Cl-stimulated H
+ currents. Interestingly, SLC4A11 appears to confer a modest amount of membrane water permeability
[15]. Several mutants show diminished water flux
[15][38][15,57]. However, in this case, water flux by wild-type SLC4A11 was shown to be inhibited by stilbenes
[15]. Further pharmacological studies examining inhibition or stimulation of SLC4A11 in the context of the different transport modes of SLC4A11 are needed.
6. SLC4A11 Plasma Membrane Function
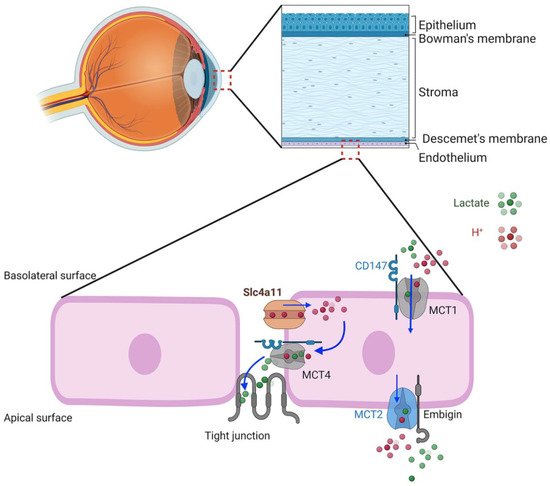
Given the properties of the SLC4A11 transporter, what might be the physiological function in corneal endothelial cells? In this regard, Parker
[34][54] and Nehrke
[39][58] have put forth a hypothesis, yet to be tested, that SLC4A11 facilitates lactate:H
+ flux and the corneal endothelium pump by actively balancing pH
i. SLC4A11 KO shows reduced lactate production and MCT4 expression in corneal endothelium, suggesting that there is some functional association
[30][32][51,52] and that SLC4A11 activity is linked to lactate transport and specifically to MCT4, which is also on the basolateral membrane. H
+ flux via SLC4A11 can proceed in either direction to balance pH
i, according to Nehrke; however the electrochemical gradient favors H
+ influx. A potential model (
Figure 2) shows H
+ influx via SLC4A11 facilitating lactate:H
+ efflux into the lateral space via MCT4 with lactate diffusion across the leaky tight junction to the apical surface. Lactate efflux via MCT4 is in line with it being high affinity and capable of lactate export in high-lactate environments
[40][59]. This lactate flux, combined with that from apical MCT2, could then provide the net lactate gradient between apical surface and basolateral space that drives water across the cells
[28][49].
Figure 2. Schematic model of corneal endothelial lactate transport. MCT1 and MCT4 require the chaperone, CD147, that targets them to the basolateral membrane. The chaperone, embigin, targets MCT2 to the apical membrane. Basolateral SLC4A11 is associated with MCT4. H+ influx driven by the negative membrane potential feeds H+ to MCT4 lactate−:H+ cotransporter, loading paracellular space with lactate that diffuses across the leaky tight junction.
7. Facilitation of Glutamine Catabolism
The apparent stimulation of SLC4A11 activity by ammonia and high pH prompts the question of physiological relevance. Ammonia is produced by cells primarily as a by-product of amino acid metabolism, particularly glutamine catabolism. Direct production of ammonia occurs in glutaminolysis, the conversion of glutamine to glutamate catalyzed by glutaminase (GLS). Glutamate can subsequently be converted to αketoglutarate by glutamate dehydrogenase (GLUD), and release a second molecule of ammonia. This occurs primarily within mitochondria, where αketoglutarate can then enter the tricarboxylic acid (TCA) cycle and contribute to the production of reducing equivalents that drive the electron transport chain, increase oxygen consumption, and produce ATP. Increases in αketoglutarate can also drive the TCA cycle in reverse, a process called reductive carboxylation. Here, the goal is to produce citrate which feeds into biosynthetic processes needed for proliferating cells. This is often found in carcinomas that express the requisite increases in TCA cycle enzymes that facilitate reductive carboxylation
[41][60]. Given the ammonia sensitivity of SLC4A11, a potential role in facilitating glutaminolysis was tested
[24][36][45,56].
Using cultured human corneal endothelial cells and mouse corneal endothelium, which highly express SLC4A11, Zhang et al. found a high expression of plasma membrane transporters for glutamine and other amino acids, GLS1 and GLUD, and a second isoform of glutaminase, GLS2, in corneal endothelial cells
[24][45]. Glucose and glutamine catabolism was then studied in the human corneal endothelial cells using stable-isotope-resolved metabolomics (SIRM), GC-MS. The TCA cycle intermediates were found to be derived from both glucose and glutamine in an approximate 50:50 ratio. Glutamine catabolism was verified by a significant production of ammonia in glutamine containing media versus glucose alone. Moreover, there was a significant increase in ATP production, suggesting that the TCA cycle is stimulated to produce more reducing equivalents. In addition, overall corneal endothelial fluid transport activity was greater in the presence of glutamine plus glucose versus glucose alone. When examining the SLC4A11 KO mouse corneal endothelium, Zhang et al.
[24][45] found increased levels of Gls1, decreased Gls2, and significantly increased levels of nitrotyrosine staining, which is commonly used as a general marker of protein oxidation related to ammonia toxicity
[42][61]. From the SLC4A11 wild-type and KO mice, Zhang et al.
[36][56] then created mouse corneal endothelial cell (MCEC) lines. They found reduced proliferative capacity, increased expression of GLS1, and a significantly reduced amount of TCA cycle intermediates derived from glutamine, suggesting that SLC4A11 is facilitating glutamine catabolism. In summary, the data from these two studies indicate that SLC4A11 facilitates the use of glutamine in the TCA cycle and reduces ammonia-related oxidative stress. In turn, the studies would predict decreased oxygen consumption and ATP production in SLC4A11 KO, which point to a dysfunction of mitochondria.
8. SLC4A11 Is a Mitochondrial Uncoupler
To examine mitochondrial function, Ogando et al.
[25][46] used SLC4A11-transfected PS120 fibroblasts, cultured human corneal endothelial cells, and SLC4A11 WT and KO mouse corneal endothelial cells (MCEC). SLC4A11 had been shown to have cytoplasmic localizations, as well as the plasma membrane
[3][43][3,42]. Ogando et al.
[25][46] found immunodetection of SLC4A11 from isolated mitochondria with an apparent inner mitochondrial membrane localization. When activated with ammonia, the mitochondrial membrane potential (MMP) depolarized, consistent with a flux of H
+ into the inner mitochondrial matrix. WT and KO cells had similar oxygen consumption rates (OCR) when incubated in glucose alone. However, glutamine significantly increased OCR in WT, but not KO. OCR analysis indicated that glutamine induced a significant proton leak in WT that was absent in KO. Glutamine caused an increase in the NAD
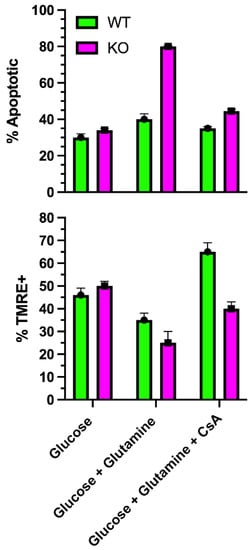
+/NADH ratio, indicating greater ETC activity, and increased ATP levels in WT, but not KO, consistent with the increased OCR. Mitochondrial reactive oxygen species (ROS) production was significantly greater in KO cells incubated in glutamine. In the absence of glutamine, mitochondrial ROS levels in KO cells were significantly lower than WT. The ROS levels paralleled the rate of apoptosis, which was highest in KO with glutamine, and lowest in WT and KO in glucose alone. The percentage of KO cells that were TMRE (tetramethylrhodamine) positive, indicating an intact MMP, dramatically decreased when incubated in glutamine, indicating ongoing mitochondrial damage. However, of the KO cells that were TMRE+ in glutamine, the staining intensity increased significantly with time, indicating MMP hyperpolarization. This suggested that glutamine catabolism in KO cells leads to increasing MMP hyperpolarization, causing increased ROS production and damage to mitochondria that then depolarizes. The damage appears to induce MTP opening (
Figure 3), which would release pro-apoptotic factors. As such, KO cells in glutamine could be rescued with the mitochondrial-directed antioxidant, MitoQ; the chemical uncoupler, BAM15; or the Gls1 inhibitors, BPTES or CB839
[25][46]. Moreover, the addition of dimethyl-αketoglutarate, which feeds the TCA cycle directly, bypassing glutaminolysis and avoiding ammonia production, reduced KO cell apoptosis, decreased mitochondrial ROS, and increased ATP levels.
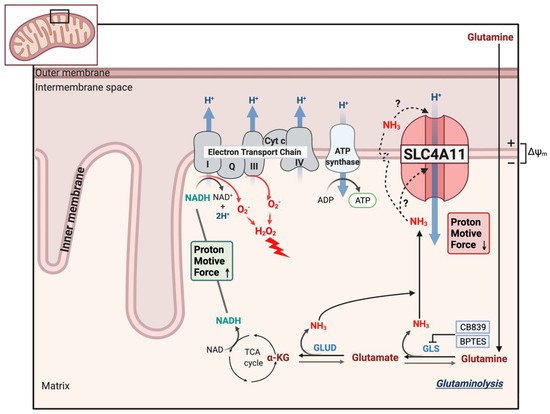
Figure 4 shows a schematic of the model of SLC4A11 mitochondrial function. Glutamine catabolism greatly accelerates TCA cycle activity, producing reducing equivalents that in turn accelerate the ETC. A by-product of complex I and III activity is superoxide production that increases with hyperpolarizing MMP
[44][45][62,63]. In addition, ammonia can have a direct catalytic effect on the complexes that increase superoxide production
[25][46][46,64]. However, SLC4A11 activated by ammonia causes H
+ influx, i.e., mitochondrial uncoupling, preventing MMP hyperpolarization and thereby reducing superoxide production.
Figure 3. Effect of glutamine on mitochondrial transition pore opening. In SLC4A11 KO mouse corneal endothelial cells, glutamine causes a significant reduction in % TMRE+ cells, (i.e., more depolarized cells), and greater % apoptosis
[25][46] (i.e., opening of MTP) that is reduced to WT levels by cyclosporine, which inhibits MTP opening (unpublished data).
Figure 4. Model of SLC4A11 mitochondrial uncoupling. Glutaminolysis feeds the TCA cycle, producing reducing equivalents that accelerate the electron transport chain, increasing the formation of superoxide (O2−) radicals that can also be exacerbated by direct action of NH3 on complex I and III. Superoxide is quickly converted to the damaging hydrogen peroxide. Ammonia either directly activates SLC4A11 or diffuses across the inner membrane and cotransports with H+ into the matrix. The mild uncoupling action (H+ influx) of SLC4A11 prevents extreme MMP hyperpolarization, excess superoxide formation, mitochondrial damage/MTP opening, and apoptosis.