Autophagy is an intracellular mechanism that maintains cellular homeostasis in different tissues. This process declines in cartilage due to aging, which is correlated with osteoarthritis (OA), a multifactorial and degenerative joint disease. Several studies show that microRNAs regulate different steps of autophagy but only a few of them participate in OA. Therefore, epigenetic modifications could represent a therapeutic opportunity during the development of OA. Besides, polyphenols are bioactive components with great potential to counteract diseases, which could reverse altered epigenetic regulation and modify autophagy in cartilage. This review aims to analyze epigenetic mechanisms that are currently associated with autophagy in OA, and to evaluate whether polyphenols are used to reverse the epigenetic alterations generated by aging in the autophagy pathway.
- Autophagy
- Osteoarthritis
- Aging
- cartilage
1. Autophagy
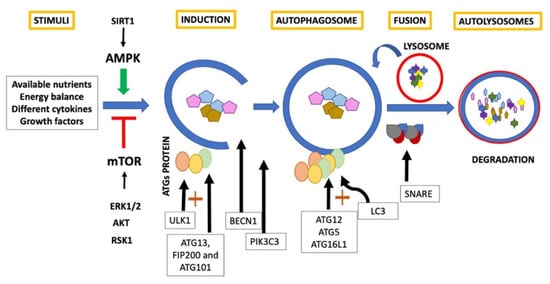
2. Autophagy and Aging
3. Autophagy and Osteoarthritis
4. Autophagy as a Therapeutic Target in OA
References
- Lotz, M.K.; Carames, B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat. Rev. Rheumatol. 2011, 7, 579–587.
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596.
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16.
- Jing, K.; Lim, K. Why is autophagy important in human diseases? Exp. Mol. Med. 2012, 44, 69–72.
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and Cellular Immune Responses. Immunity 2013, 39, 211–227.
- Füllgrabe, J.; Klionsky, D.J.; Joseph, B. The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 2013, 15, 65–74.
- Jing, Z.; Han, W.; Sui, X.; Xie, J.; Pan, H. Interaction of autophagy with microRNAs and their potential therapeutic implications in human cancers. Cancer Lett. 2015, 356, 332–338.
- Nabavi, S.F.; Sureda, A.; Dehpour, A.R.; Shirooie, S.; Silva, A.S.; Devi, K.P.; Ahmed, T.; Ishaq, N.; Hashim, R.; Sobarzo-Sánchez, E.; et al. Regulation of autophagy by polyphenols: Paving the road for treatment of neurodegeneration. Biotechnol. Adv. 2018, 36, 1768–1778.
- Carames, B.; Kiosses, W.B.; Akasaki, Y.; Brinson, D.C.; Eap, W.; Koziol, J.; Lotz, M.K. Glucosamine Activates Autophagy In Vitro and In Vivo. Arthritis Rheum. 2013, 65, 1843–1852.
- Xu, H.-D.; Wu, D.; Gu, J.-H.; Ge, J.-B.; Wu, J.-C.; Han, R.; Liang, Z.-Q.; Qin, Z.-H. The Pro-Survival Role of Autophagy Depends on Bcl-2 Under Nutrition Stress Conditions. PLoS ONE 2013, 8, e63232.
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995.
- Dong, Y.; Chen, H.; Gao, J.; Liu, Y.; Li, J.; Wang, J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol. 2019, 136, 27–41.
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Musumeci, G.; Castrogiovanni, P.; Trovato, F.M.; Weinberg, A.M.; Al-Wasiyah, M.K.; Alqahtani, M.H.; Mobasheri, A. Biomarkers of Chondrocyte Apoptosis and Autophagy in Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 20560–20575.
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699.
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215.
- Cao, Y.; Luo, Y.; Zou, J.; Ouyang, J.; Cai, Z.; Zeng, X.; Ling, H.; Zeng, T. Autophagy and its role in gastric cancer. Clin. Chim. Acta 2019, 489, 10–20.
- Legendre, F.; Ollitrault, D.; Hervieu, M.; Baugé, C.; Maneix, L.; Goux, D.; Chajra, H.; Mallein-Gerin, F.; Boumediene, K.; Galera, P.; et al. Enhanced Hyaline Cartilage Matrix Synthesis in Collagen Sponge Scaffolds by Using siRNA to Stabilize Chondrocytes Phenotype Cultured with Bone Morphogenetic Protein-2 Under Hypoxia. Tissue Eng. Part C Methods 2013, 19, 550–567.
- Zheng-Hong, Q. Autophagy: Biology and Diseases; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1206.
- Catherine, B. Regulation and Role of TGFβ Signaling Pathway in Aging and Osteoarthritis Joints. Aging Dis. 2014, 5, 394–405.
- Xu, X.; Zhi, T.; Chao, H.; Jiang, K.; Liu, Y.; Bao, Z.; Fan, L.; Wang, D.; Li, Z.; Liu, N.; et al. ERK1/2/mTOR/Stat3 pathway-mediated autophagy alleviates traumatic brain injury-induced acute lung injury. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 1663–1674.
- Mobasheri, A.; Matta, C.; Zákány, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244.
- Nikniaz, Z.; Ostadrahimi, A.; Mahdavi, R.; Ebrahimi, A.A.; Nikniaz, L. Effects of Elaeagnus angustifolia L. supplementation on serum levels of inflammatory cytokines and matrix metalloproteinases in females with knee osteoarthritis. Complement. Ther. Med. 2014, 22, 864–869.
- Wang, J.; Li, J.; Cao, N.; Li, Z.; Han, J.; Li, L. Resveratrol, an activator of SIRT1, induces protective autophagy in non-small-cell lung cancer via inhibiting Akt/mTOR and activating p38-MAPK. OncoTargets Ther. 2018, 11, 7777–7786.
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420.
- Yi, W.; Wen, Y.; Tan, F.; Liu, X.; Lan, H.; Ye, H.; Liu, B. Impact of NF-κB pathway on the apoptosis-inflammation-autophagy crosstalk in human degenerative nucleus pulposus cells. Aging 2019, 11, 7294–7306.
- Maiese, K. Moving to the Rhythm with Clock (Circadian) Genes, Autophagy, mTOR, and SIRT1 in Degenerative Disease and Cancer. Curr. Neurovascular Res. 2017, 14, 299–304.
- Pallauf, K.; Rimbach, G. Autophagy, polyphenols and healthy ageing. Ageing Res. Rev. 2013, 12, 237–252.
- Chen, K.; Yang, Y.-H.; Jiang, S.-D.; Jiang, L.-S. Decreased activity of osteocyte autophagy with aging may contribute to the bone loss in senile population. Histochem. Cell Biol. 2014, 142, 285–295.
- Zhao, G.; Zhang, W.; Li, L.; Wu, S.; Du, G. Pinocembrin Protects the Brain against Ischemia-Reperfusion Injury and Reverses the Autophagy Dysfunction in the Penumbra Area. Molecules 2014, 19, 15786–15798.
- Matsuzaki, T.; Alvarez-Garcia, O.; Mokuda, S.; Nagira, K.; Olmer, M.; Gamini, R.; Miyata, K.; Akasaki, Y.; Su, A.I.; Asahara, H.; et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci. Transl. Med. 2018, 10.
- Lapierre, L.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 11, 867–880.
- Carames, B.; Olmer, M.; Kiosses, W.B.; Lotz, M.K. The Relationship of Autophagy Defects to Cartilage Damage During Joint Aging in a Mouse Model. Arthritis Rheumatol. 2015, 67, 1568–1576.
- Weng, T.; Xie, Y.; Yi, L.; Huang, J.; Luo, F.; Du, X.; Chen, L.; Liu, C.; Chen, D. Loss of Vhl in cartilage accelerated the progression of age-associated and surgically induced murine osteoarthritis. Osteoarthr. Cartil. 2014, 22, 1197–1205.
- Van der Kraan, P. Osteoarthritis year 2012 in review: Biology. Osteoarthr. Cartil. 2012, 20, 1447–1450.
- Mobasheri, A.; Kalamegam, G.; Musumeci, G.; Batt, M.E. Chondrocyte and mesenchymal stem cell-based therapies for cartilage repair in osteoarthritis and related orthopaedic conditions. Maturitas 2014, 78, 188–198.
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759.
- Loeser, R.F. Aging processes and the development of osteoarthritis. Curr. Opin. Rheumatol. 2013, 25, 108–113.
- Musumeci, G.; Castrogiovanni, P.; Trovato, F.M.; Imbesi, R.; Giunta, S.; Szychlinska, M.A.; Loreto, C.; Castorina, S.; Mobasheri, A. Physical activity ameliorates cartilage degeneration in a rat model of aging: A study on lubricin expression. Scand. J. Med. Sci. Sports 2015, 25, e222–e230.
- De Figueroa, P.L.; Lotz, M.K.; Blanco, F.J.; Caramés, B. Autophagy Activation and Protection From Mitochondrial Dysfunction in Human Chondrocytes. Arthritis Rheumatol. 2015, 67, 966–976.
- Sasaki, H.; Takayama, K.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Fujita, N.; Oka, S.; Kurosaka, M.; Kuroda, R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2011, 64, 1920–1928.
- Duan, R.; Xie, H.; Liu, Z.-Z. The Role of Autophagy in Osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 8.
- Li, Y.; Wu, Y.; Jiang, K.; Han, W.; Zhang, J.; Xie, L.; Liu, Y.; Xiao, J.; Wang, X. Mangiferin Prevents TBHP-Induced Apoptosis and ECM Degradation in Mouse Osteoarthritic Chondrocytes via Restoring Autophagy and Ameliorates Murine Osteoarthritis. Oxidative Med. Cell. Longev. 2019, 2019, 8783197-17.
- Arias, C.; Saavedra, N.; Saavedra, K.; Alvear, M.; Cuevas, A.; Maria-Engler, S.S.; Abdalla, D.S.P.; Salazar, L.A. Propolis Reduces the Expression of Autophagy-Related Proteins in Chondrocytes under Interleukin-1β Stimulus. Int. J. Mol. Sci. 2019, 20, 3768.
- Zhang, Y.; Vasheghani, F.; Li, Y.-H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.-P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440.
- Abramoff, B.; Caldera, F.E. Osteoarthritis: Pathology, diagnosis, and treatment options. Med. Clin. N. Am. 2020, 104, 293–311.
- Skou, S.T.; Roos, E.M. Physical therapy for patients with knee and hip osteoarthritis: Supervised, active treatment is current best practice. Clin. Exp. Rheumatol. 2019, 37, 112–117.
- Wu, T.-T.; Li, W.-M.; Yao, Y.-M. Interactions between Autophagy and Inhibitory Cytokines. Int. J. Biol. Sci. 2016, 12, 884–897.
- Singh, R.; Akhtar, N.; Haqqi, T.M. Green tea polyphenol epigallocatechi3-gallate: Inflammation and arthritis. Life Sci. 2010, 86, 907–918.
- Lopez, H.L. Nutritional Interventions to Prevent and Treat Osteoarthritis. Part II: Focus on Micronutrients and Supportive Nutraceuticals. PM&R 2012, 4, S155–S168.
- Jia, G.; Cheng, G.; Gangahar, D.M.; Agrawal, D.K. Insulin-like growth factor-1 and TNF-α regulate autophagy through c- jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 2006, 84, 448–454.
- Shukla, S.; Meeran, S.M.; Katiyar, S.K. Epigenetic regulation by selected dietary phytochemicals in cancer chemoprevention. Cancer Lett. 2014, 355, 9–17.
- Imagawa, K.; De Andrés, M.C.; Hashimoto, K.; Pitt, D.; Itoi, E.; Goldring, M.B.; Roach, H.I.; Oreffo, R.O. The epigenetic effect of glucosamine and a nuclear factor-kappa B (NF-kB) inhibitor on primary human chondrocytes–implications for osteoarthritis. Biochem. Biophys. Res. Commun. 2011, 405, 362–367.
- Zhong, G.; Long, H.; Ma, S.; Shunhan, Y.; Li, J.; Yao, J. miRNA-335-5p relieves chondrocyte inflammation by activating autophagy in osteoarthritis. Life Sci. 2019, 226, 164–172.
- Lian, W.-S.; Ko, J.-Y.; Wu, R.-W.; Sun, Y.-C.; Chen, Y.-S.; Wu, S.-L.; Weng, L.-H.; Jahr, H.; Wang, F.-S. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018, 9, 919.
- Bannuru, R.R.; Osani, M.C.; Vaysbrot, E.E.; Arden, N.K.; Bennell, K.; Bierma-Zeinstra, S.M.A.; Kraus, V.B.; Lohmander, L.S.; Abbott, J.H.; Bhandari, M.; et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1578–1589.