Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Beatrix Zheng and Version 6 by Beatrix Zheng.
Chronic low-grade inflammation is involved in coronary atherosclerosis, presenting multiple clinical manifestations ranging from asymptomatic to stable angina, acute coronary syndrome, heart failure and sudden cardiac death. Coronary microvasculature consists of vessels with a diameter less than 500 μm, whose potential structural and functional abnormalities can lead to inappropriate dilatation and an inability to meet the required myocardium oxygen demands.
- coronary microvascular dysfunction
- atherosclerosis
- imaging
- biomarkers
- risk factors
- anti-inflammatory treatment
1. Introduction
Ischemic cardiac pain in the context of non-obstructed epicardial coronary arteries, a phenomenon frequently encountered in clinical cardiology practice, has been recognized as a clinical entity of increased cardiovascular risk when compared to control subjects
Ischemic cardiac pain in the context of non-obstructed epicardial coronary arteries, a phenomenon frequently encountered in clinical cardiology practice, has been recognized as a clinical entity of increased cardiovascular risk when compared to control subjects
, contrary to initial theories suggesting a nonthreatening disease progression
[3]
. Coronary microvascular dysfunction (CMVD) consists of the main etiologic factor of peripheral ischemia with “normal” epicardial coronary arteries. However, no definitive data exist on the pathophysiology, diagnosis and treatment of CMVD.
Coronary microvasculature consists of vessels with a diameter less than 500 μm, whose remodeling due to various stimuli could lead to structural and functional abnormalities and, consequently, result in inappropriate dilatation and the inability to meet the required oxygen demands. Known cardiovascular risk factors, such as diabetes mellitus and arterial hypertension, are implicated in this process due to their deleterious vascular effects. Ultimately, patients with CMVD could progress to a phenotype of heart failure with preserved ejection fraction, with variable prognosis due to a lack of disease-specific treatment
[4]
.
Chronic low-grade inflammation is undoubtedly involved in coronary atherosclerosis. Importantly, favorable cardiovascular outcomes have been reported in recent trials of patients with documented coronary artery disease (CAD) receiving either broad based or target anti-inflammatory treatment
. Contemporary evidence suggests that an inflammatory background is also responsible for the development of CMVD
[8]. Therefore, we review the latest data regarding CMVD epidemiology and diagnostic approach while elaborating on the speculated effects of inflammation and the potential therapeutic implications of immunomodulatory agents.
. Therefore, we review the latest data regarding CMVD epidemiology and diagnostic approach while elaborating on the speculated effects of inflammation and the potential therapeutic implications of immunomodulatory agents.
2. The Role of Inflammation in the Pathogenesis of CMVD
2.1. Principal Pathophysiologic Mechanisms
Evidence from studies in autoimmune rheumatic diseases have stressed the decreased nitric oxide (NO) bioavailability together with the high assembly of reactive oxygen species (ROS) as the link between the pro-inflammatory state and endothelial dysfunction observed in CMVD2.1. Principal Pathophysiologic Mechanisms
Evidence from studies in autoimmune rheumatic diseases have stressed the decreased nitric oxide (NO) bioavailability together with the high assembly of reactive oxygen species (ROS) as the link between the pro-inflammatory state and endothelial dysfunction observed in CMVD
[9]
. CMVD can be maintained by a variety of physiological processes that result in either impaired dilatation or enhanced constriction of coronary microvessels
[9]
. Following endothelial dysfunction, the ensuing inflammation and immune system dysregulation are driving forces accelerating the atherosclerotic process, consequently affecting the microvasculature. Several chemokines and cytokines are involved, with interleukin IL-1, IL-6 and TNF-α being crucial mediators of the inflammatory cascade
[10]
. Systemic inflammation is further exacerbated by pro-inflammatory circulating microparticles, which are believed to be responsible for NO modulation and cytokine release as well as monocyte recruitment
[11]
. These modifications eventually impede myocardial blood flow ability to adjust to variations in myocardial oxygen demand. Impaired vasodilation can be a result of either non-modified risk factors such as diabetes, obesity, and hypertension or either endothelium–independent mechanisms, referring to the development of nitrate resistance due to decreased cyclic GMP production
[12]
. The exacerbation of secreting oxygen species in contribution with reduced NO bioavailability drive a chain reaction of signaling events that promotes heart fibrosis and myocyte stiffness
[13]
.
The normal coronary physiology is disrupted to varied degrees as a result of CMVD. Patients with hypertrophic cardiomyopathy and those with arterial hypertension have structural abnormalities that are responsible for the development of CMVD
[14]
. In patients with non-obstructive coronary disease, dysfunction of microvasculature can be a result of several factors, such as increased heart rate, reduced diastolic time, decreased driving blood pressure, and left ventricular inotropism, which need to be considered when assessing microvascular function. Finally, the coronary blood flow linearly affected by the pulsative pattern of the heart and the intramyocardial and intraventricular pressures. As such, abnormalities in the diastolic phase of the cardiac cycle altered the myocardial perfusion
.
2.2. Hypertension
Chronic low-grade systemic inflammation seen in the cluster of comorbidities (i.e., diabetes mellitus, obesity, arterial hypertension) that are considered risk factors for CMVD merits specific attention
(
Figure 1
). Arterial hypertension increases the likelihood of death from ischemic heart disease over time. In developed countries, at least 30% of individuals have a history of hypertension, and it is independently related to poor cardiac prognosis following an acute myocardial infarction (MI)
[18]
. Severe microvascular damage inside the infarct zone presents promptly as microvascular blockage and affects approximately half of all STEMI patients. In patients with persistent microvascular obstruction, progressive irreversible capillary degradation occurs, leading to infarct zone hemorrhage, which is an independent predictor of death or heart failure long-term
. The underlying pathophysiology includes inflammatory stimuli paired with increased activation of the renin–angiotensin–aldosterone system, resulting in endothelium-dependent CMVD by recruiting cell adhesion molecules
[21]
. Oxidative stress is also central as an orchestrator or collaborator of inflammation in hypertension-related vascular aging through mediators such as osteoprotegerin and the Sphingosine Kinase 1 gene
. The increased oxidative stress could further potentiate the pro-inflammatory effects of matrix metalloproteinases (MMPs), through the promotion of a secretory endothelial cell phenotype which aids vascular aging
[24]
and, consequently, CMVD. Patients with hypertrophic cardiomyopathy and arterial hypertension have structural abnormalities that are responsible for the development of CMVD
[14]
. The morphological alterations found in each of these disorders are defined by unfavorable remodeling of intramural coronary arterioles, resulting in medial wall thickening (mostly due to smooth muscle hypertrophy and increased collagen deposition) and varying degrees of intimal thickening
[25]
.
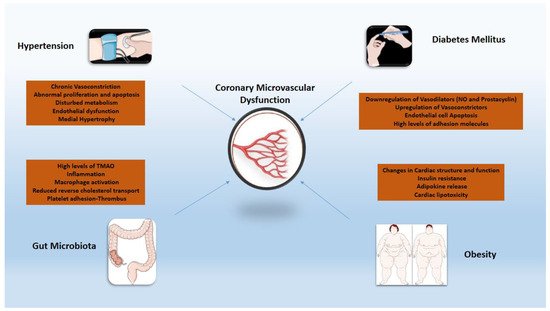
Figure 1. Inflammatory mechanisms of cardiovascular risk factors implicated in coronary microvascular dysfunction. TMAO: trimethylamine N-oxide; NO: nitric oxide.
2.3. Diabetes
In individuals with diabetes mellitus (DM), CMVD is often the prodrome of overt CAD and is a strong predictor of complications and mortality Inflammatory mechanisms of cardiovascular risk factors implicated in coronary microvascular dysfunction. TMAO: trimethylamine N-oxide; NO: nitric oxide.
2.3. Diabetes
In individuals with diabetes mellitus (DM), CMVD is often the prodrome of overt CAD and is a strong predictor of complications and mortality
. Inflammation in DM, induced by oxidative stress and the dysfunctional endothelium, plays a key role in the development of CMVD through the augmented expression of adhesion molecules and inflammatory cytokines as well as the mobilization of vascular smooth muscle cells
. Vascular adhesion protein-1 (VAP-1), another molecule involved in the pathophysiology of DM, is also overexpressed by damaged endothelial cells, affecting leukocyte recruitment and advanced glycation end products (AGE) generation, thus promoting a pro-inflammatory state
. Chemokines such as CC-chemokine ligand 2–3 and CXC-chemokine ligand 8 have also been found to be involved in leukocyte mobilization and macrophage recruitment, resulting in an augmented inflammatory response in the setting of DM
[34]
. TNF-α values correlated with the levels of glucose and cystatin C, confirming that diabetes plays an orchestrating role in the progress of CMVD
[35]
. Coronary microvascular dysfunction has an independent relationship with cardiac or all-cause mortality in people with and without diabetes, allowing for incremental risk classification. Patients with diabetes and impaired CFR have by far more increased risk for cardiac events than those with normal CFR
[36]
. Importantly, studies have stressed the importance of strict glycemic control in maintaining vascular integrity
, a finding that could be partly justified by attenuation of the above mentioned pro-inflammatory, DM-induced effects via optimal management
.
2.4. Obesity
Weight status constitutes a significant role in the progression of atherosclerotic disease
[39]
. Obesity is characterized by excessive expansion of visceral white adipose tissue mass, also known as adiposopathy. Adiposopathy is comprised of adipocyte hypertrophy, decreased adipose tissue blood flow, altered oxygen levels within the tissue, a state of chronic low-grade inflammation and blunted lipid metabolism
. The latter includes impaired capacity for storing the surplus of dietary lipids, resulting in deposition of ectopic fat accumulating in body locations where it is not physiologically stored, such as the liver and muscle, and a shift to visceral adipose tissue (fat storage in the intraperitoneal and retroperitoneal spaces), contributing to increased circulating free fatty acids, oxidative stress, systemic inflammation, adipokine dysregulation and insulin resistance
. However, epicardial adipose tissue has several unique properties that distinguish it from other depots of visceral fat due to the common microcirculation of epicardial tissue and the underlying myocardium
[46]
. The accumulation of epicardial fat is closely associated with an impaired myocardial microcirculation, cardiac diastolic filling abnormalities, increased vascular stiffness, and left atrial dilatation in obese people
. More specifically, high levels of TNF-α induce the secretion of adhesion and chemoattractant molecules such as vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and monocyte chemoattractant protein-1 while reducing nitric oxide availability. Resistin hormone stimulates the proliferation of smooth muscle cells and the over-secretion of endotelin-1, leading to endothelial dysfunction
[49]
. Obesity-related reductions in myocardial blood flow due to CMVD, combined with increased cardiac metabolic demand due to increases in ventricular mass, volume expansion, higher filling pressures, and greater cardiac output, may create a perfect background for the occurrence of myocardial oxygen supply–demand mismatch
[36]
. Finally, an unfavorable correlation was observed between CFR and increased LDL cholesterol where treatment with pioglitazone and lipid-lowering therapy seemed to improve CFR
[50].
2.5. Gut Microbiota
This term describes the various commensal microbial species in the gastrointestinal tract.
2.5. Gut Microbiota
This term describes the various commensal microbial species in the gastrointestinal tract
[51]
. During the last decade, several studies reported the potential association between gut microbiota and atherosclerosis
. Choline, betaine and L-carnitine are metabolized to trimethylamine (TMA) which is generated to trimethylamine N-oxide (TMAO), a gut microbe-dependent metabolite
. Studies showed that increased TMAO level induced the activation of NF-kappa B (NF-κB) pathway and increased the expression of pro-inflammatory genes including inflammatory cytokines, adhesion molecules and chemokines
[53]
. Oxidative stress and NOD-like receptor protein 3 (NLRP3) inflammasome activation could also be triggered by TMAO and inflammatory cytokines such as IL-18 and IL-1β released. As such, diet has an important role in affecting the concentration of TMAO levels and the progression of atherosclerosis
. Latest studies showed that the use of broad-spectrum antibiotics for 3 to 4 weeks suppressed TMAO levels, ameliorating age-related oxidative stress and arterial dysfunction in mice
[56]
. The ability of CAD patients’ microbiota to generate ‘secondary’ bile acids enhanced the variety of the bile acid pool in both feces and serum. Under a high fat diet, this mechanism impeded hepatic bile acid synthesis and resulted in higher blood cholesterol levels. The CAD microbiota enhanced circulatory lipopolysaccharides levels as well as pro-inflammatory cytokine expression, while activating intestinal and systemic T help responses and decreasing Treg cell dispersion
[57]
. Finally, the use of probiotics, synbiotics, and probiotic functional products in a study of 90 obese patients with CAD proved beneficial, controlling plasma TMAO and HDL-C levels
[58]. Further studies are needed to evaluate the efficacy of inhibiting various steps of TMAO production for the management atherosclerotic disease.
. Further studies are needed to evaluate the efficacy of inhibiting various steps of TMAO production for the management atherosclerotic disease.
References
- Johnson, B.D.; Shaw, L.J.; Buchthal, S.D.; Bairey Merz, C.N.; Kim, H.W.; Scott, K.N.; Doyle, M.; Olson, M.B.; Pepine, C.J.; den Hollander, J.; et al. Prognosis in women with myocardial ischemia in the absence of obstructive coronary disease: Results from the National Institutes of Health-National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation 2004, 109, 2993–2999.
- Lin, F.Y.; Shaw, L.J.; Dunning, A.M.; Labounty, T.M.; Choi, J.H.; Weinsaft, J.W.; Koduru, S.; Gomez, M.J.; Delago, A.J.; Callister, T.Q.; et al. Mortality risk in symptomatic patients with nonobstructive coronary artery disease: A prospective 2-center study of 2583 patients undergoing 64-detector row coronary computed tomographic angiography. J. Am. Coll. Cardiol. 2011, 58, 510–519.
- Lichtlen, P.R.; Bargheer, K.; Wenzlaff, P. Long-term prognosis of patients with anginalike chest pain and normal coronary angiographic findings. J. Am. Coll. Cardiol. 1995, 25, 1013–1018.
- Elgendy, I.Y.; Pepine, C.J. Heart Failure With Preserved Ejection Fraction: Is Ischemia Due to Coronary Microvascular Dysfunction a Mechanistic Factor? Am. J. Med. 2019, 132, 692–697.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505.
- Sagris, M.; Antonopoulos, A.S.; Theofilis, P.; Oikonomou, E.; Siasos, G.; Tsalamandris, S.; Antoniades, C.; Brilakis, E.S.; Kaski, J.C.; Tousoulis, D. Risk factors profile of young and older patients with Myocardial Infarction. Cardiovasc. Res. 2021, cvab264.
- Zanatta, E.; Colombo, C.; D’Amico, G.; d’Humieres, T.; Dal Lin, C.; Tona, F. Inflammation and Coronary Microvascular Dysfunction in Autoimmune Rheumatic Diseases. Int. J. Mol. Sci. 2019, 20, 5563.
- Faccini, A.; Kaski, J.C.; Camici, P.G. Coronary microvascular dysfunction in chronic inflammatory rheumatoid diseases. Eur. Heart J. 2016, 37, 1799–1806.
- Oikonomou, E.; Leopoulou, M.; Theofilis, P.; Antonopoulos, A.S.; Siasos, G.; Latsios, G.; Mystakidi, V.C.; Antoniades, C.; Tousoulis, D. A link between inflammation and thrombosis in atherosclerotic cardiovascular diseases: Clinical and therapeutic implications. Atherosclerosis 2020, 309, 16–26.
- Puddu, P.; Puddu, G.M.; Cravero, E.; Muscari, S.; Muscari, A. The involvement of circulating microparticles in inflammation, coagulation and cardiovascular diseases. Can. J. Cardiol. 2010, 26, 140–145.
- Duncker, D.J.; Bache, R.J. Regulation of coronary blood flow during exercise. Physiol. Rev. 2008, 88, 1009–1086.
- Clarke, J.G.; Davies, G.J.; Kerwin, R.; Hackett, D.; Larkin, S.; Dawbarn, D.; Lee, Y.; Bloom, S.R.; Yacoub, M.; Maseri, A. Coronary artery infusion of neuropeptide Y in patients with angina pectoris. Lancet 1987, 1, 1057–1059.
- Crea, F.; Camici, P.G.; Bairey Merz, C.N. Coronary microvascular dysfunction: An update. Eur. Heart J. 2014, 35, 1101–1111.
- Choudhury, L.; Rosen, S.D.; Patel, D.; Nihoyannopoulos, P.; Camici, P.G. Coronary vasodilator reserve in primary and secondary left ventricular hypertrophy. A study with positron emission tomography. Eur. Heart J. 1997, 18, 108–116.
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Tsioufis, C.; Oikonomou, E.; Antoniades, C.; Crea, F.; Kaski, J.C.; Tousoulis, D. Inflammatory Mechanisms in COVID-19 and Atherosclerosis: Current Pharmaceutical Perspectives. Int. J. Mol. Sci. 2021, 22, 6607.
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781.
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603.
- Carrick, D.; Haig, C.; Ahmed, N.; McEntegart, M.; Petrie, M.C.; Eteiba, H.; Hood, S.; Watkins, S.; Lindsay, M.M.; Davie, A.; et al. Myocardial Hemorrhage After Acute Reperfused ST-Segment-Elevation Myocardial Infarction: Relation to Microvascular Obstruction and Prognostic Significance. Circ. Cardiovasc. Imaging 2016, 9, e004148.
- Eitel, I.; Kubusch, K.; Strohm, O.; Desch, S.; Mikami, Y.; de Waha, S.; Gutberlet, M.; Schuler, G.; Friedrich, M.G.; Thiele, H. Prognostic value and determinants of a hypointense infarct core in T2-weighted cardiac magnetic resonance in acute reperfused ST-elevation-myocardial infarction. Circ. Cardiovasc. Imaging 2011, 4, 354–362.
- Duprez, D.A. Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: A clinical review. J. Hypertens. 2006, 24, 983–991.
- Hao, Y.; Tsuruda, T.; Sekita-Hatakeyama, Y.; Kurogi, S.; Kubo, K.; Sakamoto, S.; Nakamura, M.; Udagawa, N.; Sekimoto, T.; Hatakeyama, K.; et al. Cardiac hypertrophy is exacerbated in aged mice lacking the osteoprotegerin gene. Cardiovasc. Res. 2016, 110, 62–72.
- Siedlinski, M.; Nosalski, R.; Szczepaniak, P.; Ludwig-Galezowska, A.H.; Mikolajczyk, T.; Filip, M.; Osmenda, G.; Wilk, G.; Nowak, M.; Wolkow, P.; et al. Vascular transcriptome profiling identifies Sphingosine kinase 1 as a modulator of angiotensin II-induced vascular dysfunction. Sci. Rep. 2017, 7, 44131.
- Wang, M.; Kim, S.H.; Monticone, R.E.; Lakatta, E.G. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension 2015, 65, 698–703.
- Camici, P.G.; Olivotto, I.; Rimoldi, O.E. The coronary circulation and blood flow in left ventricular hypertrophy. J. Mol. Cell Cardiol. 2012, 52, 857–864.
- Cortigiani, L.; Rigo, F.; Gherardi, S.; Galderisi, M.; Bovenzi, F.; Sicari, R. Prognostic meaning of coronary microvascular disease in type 2 diabetes mellitus: A transthoracic Doppler echocardiographic study. J. Am. Soc. Echocardiogr. 2014, 27, 742–748.
- Murthy, V.L.; Naya, M.; Foster, C.R.; Gaber, M.; Hainer, J.; Klein, J.; Dorbala, S.; Blankstein, R.; Di Carli, M.F. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 2012, 126, 1858–1868.
- Sagris, M.; Giannopoulos, S.; Giannopoulos, S.; Tzoumas, A.; Texakalidis, P.; Charisis, N.; Kokkinidis, D.G.; Malgor, R.D.; Mouawad, N.J.; Bakoyiannis, C. Transcervical carotid artery revascularization: A systematic review and meta-analysis of outcomes. J. Vasc. Surg. 2021, 74, 657–665.e612.
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74.
- Suzuki, L.A.; Poot, M.; Gerrity, R.G.; Bornfeldt, K.E. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: Lack of direct growth-promoting effects of high glucose levels. Diabetes 2001, 50, 851–860.
- Salmi, M.; Jalkanen, S. Vascular Adhesion Protein-1: A Cell Surface Amine Oxidase in Translation. Antioxid. Redox Signal. 2019, 30, 314–332.
- Stolen, C.M.; Madanat, R.; Marti, L.; Kari, S.; Yegutkin, G.G.; Sariola, H.; Zorzano, A.; Jalkanen, S. Semicarbazide sensitive amine oxidase overexpression has dual consequences: Insulin mimicry and diabetes-like complications. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 702–704.
- Diavati, S.; Sagris, M.; Terentes-Printzios, D.; Vlachopoulos, C. Anticoagulation Treatment in Venous Thromboembolism: Options and Optimal Duration. Curr. Pharm. Des. 2021.
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107.
- Sorop, O.; Heinonen, I.; van Kranenburg, M.; van de Wouw, J.; de Beer, V.J.; Nguyen, I.T.N.; Octavia, Y.; van Duin, R.W.B.; Stam, K.; van Geuns, R.J.; et al. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc. Res. 2018, 114, 954–964.
- Camici, P.G.; d’Amati, G.; Rimoldi, O. Coronary microvascular dysfunction: Mechanisms and functional assessment. Nat. Rev. Cardiol. 2015, 12, 48–62.
- Costantino, S.; Paneni, F.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Tanese, L.; Russo, G.; Pitocco, D.; Lanza, G.A.; et al. Impact of Glycemic Variability on Chromatin Remodeling, Oxidative Stress, and Endothelial Dysfunction in Patients With Type 2 Diabetes and With Target HbA1c Levels. Diabetes 2017, 66, 2472–2482.
- Siasos, G.; Skotsimara, G.; Oikonomou, E.; Sagris, M.; Vasiliki-Chara, M.; Bletsa, E.; Stampouloglou, P.; Theofilis, P.; Charalampous, G.; Tousoulis, D. Antithrombotic Treatment in Diabetes Mellitus: A Review of the Literature about Antiplatelet and Anticoagulation Strategies Used for Diabetic Patients in Primary and Secondary Prevention. Curr. Pharm. Des. 2020, 26, 2780–2788.
- Neeland, I.J.; Ross, R.; Despres, J.P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: A position statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725.
- Shulman, G.I. Ectopic Fat in Insulin Resistance, Dyslipidemia, and Cardiometabolic Disease. N. Engl. J. Med. 2014, 371, 1131–1141.
- Goossens, G.H.; Blaak, E.E. Adipose Tissue Dysfunction and Impaired Metabolic Health in Human Obesity: A Matter of Oxygen? Front. Endocrinol. 2015, 6, 55.
- Frayn, K.N.; Karpe, F. Regulation of human subcutaneous adipose tissue blood flow. Int. J. Obes. 2014, 38, 1019–1026.
- Lempesis, I.G.; van Meijel, R.L.J.; Manolopoulos, K.N.; Goossens, G.H. Oxygenation of adipose tissue: A human perspective. Acta Physiol. 2019, 228, e13298.
- Bays, H.E. Adiposopathy is “sick fat” a cardiovascular disease? J. Am. Coll. Cardiol. 2011, 57, 2461–2473.
- Bays, H.E. Adiposopathy, diabetes mellitus, and primary prevention of atherosclerotic coronary artery disease: Treating “sick fat” through improving fat function with antidiabetes therapies. Am. J. Cardiol. 2012, 110, 4B–12B.
- Cao, H. Adipocytokines in obesity and metabolic disease. J. Endocrinol. 2014, 220, T47–T59.
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 120.
- Sagris, M.; Kokkinidis, D.G.; Lempesis, I.G.; Giannopoulos, S.; Rallidis, L.; Mena-Hurtado, C.; Bakoyiannis, C. Nutrition, dietary habits, and weight management to prevent and treat patients with peripheral artery disease. Rev. Cardiovasc. Med. 2020, 21, 565–575.
- Packer, M. Epicardial Adipose Tissue May Mediate Deleterious Effects of Obesity and Inflammation on the Myocardium. J. Am. Coll. Cardiol. 2018, 71, 2360–2372.
- Naoumova, R.P.; Kindler, H.; Leccisotti, L.; Mongillo, M.; Khan, M.T.; Neuwirth, C.; Seed, M.; Holvoet, P.; Betteridge, J.; Camici, P.G. Pioglitazone improves myocardial blood flow and glucose utilization in nondiabetic patients with combined hyperlipidemia: A randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol. 2007, 50, 2051–2058.
- Lang, D.H.; Yeung, C.K.; Peter, R.M.; Ibarra, C.; Gasser, R.; Itagaki, K.; Philpot, R.M.; Rettie, A.E. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: Selective catalysis by FMO3. Biochem. Pharmacol. 1998, 56, 1005–1012.
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143.
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37.
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126.
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J. Am. Heart Assoc. 2016, 5, e002767.
- Brunt, V.E.; Gioscia-Ryan, R.A.; Richey, J.J.; Zigler, M.C.; Cuevas, L.M.; Gonzalez, A.; Vazquez-Baeza, Y.; Battson, M.L.; Smithson, A.T.; Gilley, A.D.; et al. Suppression of the gut microbiome ameliorates age-related arterial dysfunction and oxidative stress in mice. J. Physiol. 2019, 597, 2361–2378.
- Liu, H.; Tian, R.; Wang, H.; Feng, S.; Li, H.; Xiao, Y.; Luan, X.; Zhang, Z.; Shi, N.; Niu, H.; et al. Gut microbiota from coronary artery disease patients contributes to vascular dysfunction in mice by regulating bile acid metabolism and immune activation. J. Transl. Med. 2020, 18, 382.
- Tenore, G.C.; Caruso, D.; Buonomo, G.; D’Avino, M.; Ciampaglia, R.; Maisto, M.; Schisano, C.; Bocchino, B.; Novellino, E. Lactofermented Annurca Apple Puree as a Functional Food Indicated for the Control of Plasma Lipid and Oxidative Amine Levels: Results from a Randomised Clinical Trial. Nutrients 2019, 11, 122.
More