Neurodegenerative diseases are debilitating and currently incurable conditions causing severe cognitive and motor impairments, defined by the progressive deterioration of neuronal structure and function, eventually causing neuronal loss. Understand the molecular and cellular mechanisms underlying these disorders are essential to develop therapeutic approaches. MicroRNAs (miRNAs) are short non-coding RNAs implicated in gene expression regulation at the post-transcriptional level. Moreover, miRNAs are crucial for different processes, including cell growth, signal transmission, apoptosis, cancer and aging-related neurodegenerative diseases. Altered miRNAs levels have been associated with the formation of reactive oxygen species (ROS) and mitochondrial dysfunction. Mitochondrial dysfunction and ROS formation occur in many neurodegenerative diseases such as Alzheimer’s, Parkinson’s and Huntington’s diseases. The crosstalk existing among oxidative stress, mitochondrial dysfunction and miRNAs dysregulation plays a pivotal role in the onset and progression of neurodegenerative diseases.
- miRNA, neurodegeneration, potential treatment
1. miRNA Dysregulation and Mitochondrial Dysfunction in Alzheimer’s
AD is one of the most prevalent diseases that affect the aged people, characterized by memory loss, impaired cognitive function and various neuropsychiatric disturbances. Two varieties of AD exist, sporadic and familial. Early-onset AD, recognized as familial AD, is a sporadic form of the disease observed in 1–2% of all AD cases. Numerous studies reported some biochemical and genetic alterations that participate to the onset, progressive degeneration and neuronal death in AD [1]: deposition of amyloid β (Aβ) peptide, proteasome inhibition [2], oxidative stress [3], mitochondrial dysfunction [4], hyperphosphorylation of tau protein and heritable mutations in presenilin 1, presenilin 2 [5] and Aβ precursor protein (APP) genes [6] (illustrated in Figure 1).
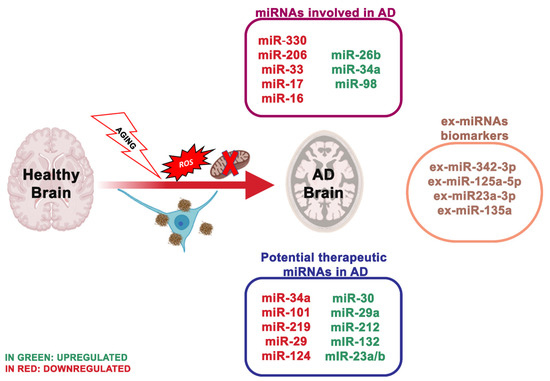
Figure 1. The most essential miRNAs involved in Alzheimer’s disease.
Compromised energy uptake and defects in mitochondrial respiration are essential characteristics of brain tissue altered by neurodegeneration, but also of peripheral cells (platelets and fibroblasts) in AD patients. Specifically, decreased cerebral metabolism has been shown in the temporoparietal cortices of AD patients, and these variations lead to both neuropsychological impairment and atrophy, as demonstrated by neuroimaging analyses [7][99]. In the last years, numerous researchers tried to understand the underlying mechanism of brain metabolism decline during AD and to identify a possible connection of the reductions in mitochondrial enzyme activities with premorbid cognitive level and with plaque counts [8][100].
Variations in mtDNA levels, typically assessed as the mitochondrial genome to the nuclear genome ratio and the mtDNA levels in body tissues and fluids, represent a biomarker of mitochondrial aberration [8]. Chen et al. 2018 [9] examined, in scopolamine-treated mice, an animal model of AD, the effect of miR-98 on Aβ-protein, oxidative stress and altered mitochondria activity via the Notch signaling pathway by targeting hairy and enhancer of split (Hes)-related with YRPW motif protein 2 (HEY2). The data obtained in humans revealed that the expression of miR-98 was significantly different in patients with AD respect with the controls; in the AD group, when miR-98 was lower, and the expression of HEY2 was elevated, the Notch-HEY2 pathway was stimulated [10]. The Notch1 pathway is a cellular cascade with primary roles in brain development and the adult brain; the increased activation of this pathway following brain damage is deleterious for neuronal survival [11]. MiR-98 targeting HEY2 prevented the activity of the Notch pathway, promoting to the inhibition of the production of Aβ and the increase of oxidative stress and mitochondria dysfunction in AD mice. An earlier study showed that miR-98-5p regulated the expression of Sorting Nexin 6 (SNX6) and was crucial for Aβ deposits [12]. miR-98 also led to AD-like disorder by targeting insulin-like growth factor 1 and, in turn, supporting the production of Aβ, thus implying that miR-98 is vital in the development of the pathology of AD [13]. In the sight of this, additional studies are necessary to validate the impacts of miR-98 in the regulation of AD mice by targeting HEY2 through the Notch signaling pathway before its consideration as an appropriate treatment for AD. Zhou et al. [14][
50] described the effect of miRNA targeting proto-oncogene vav (VAV) on Aβ production, oxidative stress and mitochondrial malfunction in AD mice through the MAPK signaling pathway. [14]
MiR-330 appeared decreased in neuronal cells of AD mice, and proto-oncogene VAV1 was negatively controlled by miR-330. In contrast with the healthy animals, the positive protein expression rate of VAV1 was considerably higher in the AD group. Increased level of miR-330 reduced the expression of VAV1, c-Jun N-terminal kinase (JNK1), extracellular signaling kinase 1 (ERK1), mitogen-activated protein kinase (P38) and Aβ. Still, it enhanced the expression of low-density lipoprotein receptor-related protein-1 (LRP-1) and cyclooxygenase [15]. AD mice showed high Aβ production with reduced Cu/Zn Super Oxide Dismutase 1 (SOD1) levels. Moreover, miR-195 was related to mitochondrial dysfunction by mitofusin 2 deregulation. Thus, the overexpression of miR-330 in AD supports the oxidative stress, and mitochondrial dysfunction by targeting VAV1 through the MAPK signaling pathway. Another research group reported that miR-30 family members prevent mitochondrial fission targeting p53, which stimulates mitochondrial fission by transcriptionally upregulating dynamin-related protein 1 (Drp1) expression [16]. Further, miRNAs represent potential, non-invasive peripheral biomarkers in aging and other age-related disorders, including AD [17]; in fact, several miRNAs [18] were strongly influenced by age either before or during Aβ plaque deposits.
The latest studies regarding miRNAs as biomarkers are moving towards the use of ex-miRNAs [19]. Ex-miRNAs are more stable, more reliable and change upon aging, diets and pathologic states. Lugli and collaborators [20], isolated exosomes from AD patients plasma and healthy group to analyze miRNA expression using high-throughput sequencing technology. Notably, ex-miR-342-3p levels were substantially reduced in AD patients. These data were also confirmed by Rani et al. [21]; they also revealed lower levels of ex-miR-125a-5p, ex-miR-125b5p and ex-miR-451a levels in AD, and the reduction of these ex-miRNAs is associated with the extent of cognitive impairment. Another ex-miRNA study, including 10 AD patients and 15 controls, through next-generation sequencing technology, identified reduced levels of ex-miR23a-3p, ex-let-7i-5p, ex-miR-126-3p and ex-miR-151a-3p in AD patients; indicating that these altered ex-miRNAs levels in the plasma showed diagnostic value for AD [22]. Yang et al. [23] through quantitative RT-PCR measured the serum levels of three ex-miRNAs, ex-miR-135a, ex-miR-193b and ex-miR-384 in 101 patients with mild cognitive impairment and 107 patients with dementia of Alzheimer’s type (DAT). The levels of ex-miR-135a and ex-miR-384 were increased in DAT patients, while ex-miR-193b resulted reduced. Furthermore, the combination of the three miRNAs would be better for the early diagnosis of AD than any single one.
2. miRNA Dysregulation and Mitochondrial Dysfunction in Parkinson
2.1. miRNA Dysregulation and Mitochondrial Dysfunction in Parkinson
PD is the second most frequent neurodegenerative disorder concerning 1% of the population over 55 years [24]. It is clinically described by motor symptoms, involving resting tremor, muscle stiffness, bradykinesia and postural instability. In PD, the imbalance between mitochondrial dysfunction and ROS production results in oxidative stress and neuronal degeneration. In neuronal cells, ROS can injure macromolecules, comprising of nucleic acids, lipids and proteins, inducing dopaminergic (DA) neuron degeneration and neuronal network alteration, eventually leading to PD [25]. It is well known that oxidative stress is involved in PD. There is fascination in identifying how miRNAs are interested in the pathological processes and how they are involved in oxidative stress, mitochondrial dysfunction [26], α-synuclein aggregation [27], neuroinflammation [28] and dysregulation of the endogenous antioxidant system [29] (
Figure 2). miRNAs control the gene expression in the cortex of PD patients, and the cortical expression patterns of miRNAs accurately discern PD from healthy brains [30]. Six miRNAs are correlated with the dopaminergic phenotype in PD [31]:
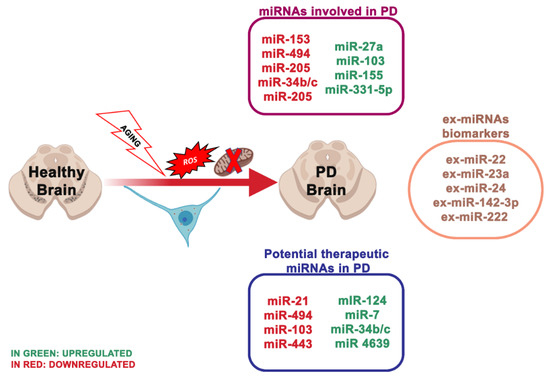
Figure 2.
-
miR-133b that controls the transcriptional activator Pitx3, which is a crucial component in the development of the DA neuronal phenotype in vivo [32];
miR-133b that controls the transcriptional activator Pitx3, which is a crucial component in the development of the DA neuronal phenotype in vivo [74]; -
miR-7, which suppressed α-synuclein in human neuroblastoma cells, and it may be inhibited by oxidative stress in vitro and in vivo [33].
miR-7, which suppressed α-synuclein in human neuroblastoma cells, and it may be inhibited by oxidative stress in vitro and in vivo [53]. -
miR153, conserved across vertebrate species, and inhibited from α-synuclein [34];
-
miR-433, related to a mutation of its binding region in the 3′UTR region of the FGF20 gene. miR-433 prevented the translation of the FGF 20 gene in vitro. Single nucleotide polymorphism in FGF20 shows that the genetic variability of FGF20 may represent a PD risk [35].
miR-433, related to a mutation of its binding region in the 3′UTR region of the FGF20 gene. miR-433 prevented the translation of the FGF 20 gene in vitro. Single nucleotide polymorphism in FGF20 shows that the genetic variability of FGF20 may represent a PD risk [56]. -
miR-205, transfecting miR-205 in the neurons expressing a PD-related LRKK2 R1441G mutant avoided the neural development defects [36].
miR-205, transfecting miR-205 in the neurons expressing a PD-related LRKK2 R1441G mutant avoided the neural development defects [123]. -
miR-124 controls the transcription activator FoxA2, relevant in midbrain dopaminergic cell development in both rodents and humans [37], and its role is necessary for dopaminergic neurons survival in PD mice [38].
Some of these miRNAs described are also implicated in the destruction of mitochondrial homeostasis. Once mitochondrial dysfunction occurs, leakage of ETC may directly stimulate ROS production, thus intensifying the neuronal impairment. Mitochondrial impairment and decreased complex I activity were detected in the substantia nigra pars compacta (SNpc) and frontal cortex of PD patients [39]. Wang et al. [40] demonstrated that miR-124 regulates the apoptosis and autophagy process in the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) model of PD. In contrast, elevated miR-124 levels impede the expression of the protein bcl-2-like protein 11 (Bim), reducing the translocation of its downstream protein bcl-2-like protein 4 (Bax) to mitochondria and lysosome, consequently inhibiting mitochondria apoptotic signaling pathways and stabilizing the impaired autophagic activity.
The encoding gene of protein DJ-1 (PARK-7) can induce autosomal recessive PD with reduced DJ-1 levels in the SNpc. Several investigations revealed that DJ-1 could directly combine to the subunits of complex I and sustain its function as an integral mitochondrial protein [41]. Mitochondrial morphology and dynamics are altered in DJ-1-knockdown neurons [42]. An exciting study suggested that miR-494 may exacerbate oxidative stress-induced neuronal damage by reducing DJ-1 expression [43]. Another study suggested that decreased miR34b/c levels parallel with reduced expression of DJ-1 impact to mitochondrial impairment in PD patients’ brains, confirmed in miR-34b/c-depleted cells. MiR-34b/c may regulate DJ-1 expression in an indirect manner; however, the specific mechanism is still unclear [44]. Another protein of interest in mitochondrial dynamics is leucine-rich repeat kinase 2 (LRRK2). Increased LRRK2 protein levels can alter the mitochondrial dynamics and integrity via dynamin-like protein (DLP1), and, notably, miR-205 expression is significantly reduced in PD patient brains, parallel with higher LRRK2 protein levels [45][46]. Further studies showed that miR-205 inhibits LRRK2 protein expression in primary neurons by targeting the 3′-UTR of the LRRK2 gene [47]. SH-SY5Y cells, upon tumor necrosis factor-α treatment, showed improved miR-27a and miR-103 levels, that may inhibit the expression of the functional units of complex I. Elevated levels of miR-155 and miR-27a may lead to mitochondrial dysfunction and oxidative stress, reducing the transcript levels of the ATP synthase membrane subunit c locus 3 (ATP5G3), a subunit of complex V [48].
Additionally, miR-7 stabilizes mitochondrial membrane potential by inhibiting the expression of the voltage-dependent anion channel 1 (VDAC1), one element of the mitochondrial permeability transition pore, which may represent a possible target for decreasing the mitochondrial dysfunction in PD [49]. Another essential protein involved in mitochondrial homeostasis is the peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1α), a crucial activator for mitochondrial genes, reduced in PD patients [50].
In consideration of the vital role of miRNAs in PD pathogenesis and progression, small RNA molecules seem an encouraging means in PD therapy, leading to potential molecular targets and supporting improved and personalized therapeutic approaches [51]. Valdes et al. [52] developed small double-stranded RNA molecules that mimic miRNAs and can function as gain-of-function tools for specific miRNAs. This miRNA can mimic reduced target proteins by interacting with the 3′UTR of the mRNA of a specific target gene. Thus, they can regulate PD-related risk genes and proteins associated with PD. However, this strategy may have possible risks of off-target effects and the possibility of undesirable interfering with other genes.
3. miRNA Dysregulation and Mitochondrial Dysfunction in Huntington’s Disease
2.2. miRNA Dysregulation and Mitochondrial Dysfunction in Huntington’s Disease
HD is a dominantly inherited neurodegenerative disease clinically described by cognitive impairment, gradual movement condition and psychiatric problems [53]. The main characteristic of HD is the striatal medium spiny neurons (MSNs) degeneration, but also of deep-layer cortical pyramidal neurons. HD is due to the CAG trinucleotide repeat expansion encoding an extended polyglutamine (polyQ) section, adjacent to the N-terminus of Huntingtin (HTT) [54] (
Figure 3). Healthy people have CAG repeat lengths that vary between 6 and 35, while HD patients show repeat lengths superior to 36 on one HTT allele, with the length of the CAG repeats inversely related to the age of HD onset [55]. To date, there is no cure to counteract the onset or advancement of HD.
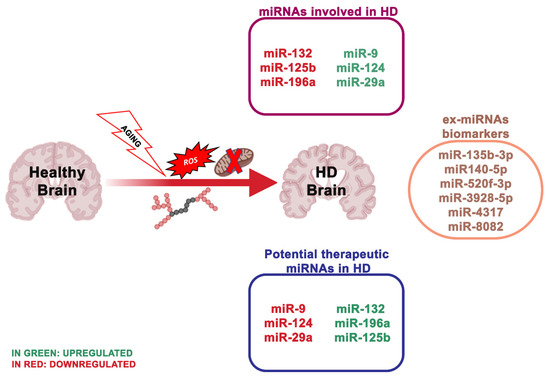
Figure 3.
How altered mitochondria and bioenergetic impairment in HD developed is still unclear [56][
146] [57][58][59], supporting that mHTT directly led to mitochondrial dysfunction. In addition, this relation is due to the capability of mHTT to reduce the expression of a key protein of mitochondrial homeostasis, PGC-1α [60][
147]. mHTT inhibits the PGC-1α pathway, which in turn avoids the activation of downstream pathways, whereas PGC-1α ectopic expression resulted neuroprotective in transgenic HD mice and the 3-NPA mouse model [61]. The upregulation and downregulation of some neuroactive miRNAs can influence mitochondrial homeostasis through the regulation of PGC-1. We can, therefore, hypothesize that by regulating the miRNAs implicated in mitochondrial dysfunctions, we could indirectly act on the interaction between mHTT and OMM, which appears to be the basis of mitochondrial dysfunctions in the HD.
Numerous researchers are focused on the study of therapies able to lower the HTT expression in the brain. The study by Stanek et al. [62] offers additional proof regarding the importance of astrocytes in the HD pathology. Indeed, astrocytes are fundamental for neural circuits; however, it is still unknown if they participate in the mechanism to initiate HD. Increasing studies reported that the presence of mHTT in astrocytes induces reduced expression of glutamate transporters and aberrant glutamate uptake, which is sufficient to led neurodegeneration in medium spiny neurons of the striatum [63]. Impaired neuronal excitability and excitotoxicity linked with HD may, therefore, be a result of altered astrocyte function [64].
MiRNAs deregulation has been described in HD in vitro models, transgenic HD animals and human HD brains [65][66]. The study of Langfelder et al. [67] was the first to explain Htt-CAG length-dependent alterations in miRNA expression in the brain regions differentially susceptible to HD. In particular, a high number (
n
n
n
n = 45). Notably, the number of deregulated miRNAs in the cerebellum was double that in the cortex and hippocampus, even though the cerebellum is fairly unaffected in HD. A larger study evaluated miRNA expression in HD patients, revealing multiple differentially expressed miRNAs, some of which modulating neuron survival [68]. The results obtained by Hoss et al.[69] indicated numerous miRNA changes in the HD brain, and most of these are associated with clinical manifestations of HD, where the signal is impartial of the size of the CAG repeat extension. Other research used a next-generation sequence analysis of small non-coding RNAs to analyze 26 HD patients and 36 healthy individuals. Nine hundred and thirty eight miRNAs were found, and 75 of these were differentially expressed. Concordant with these results, the downregulation of miR-132-3p in human HD parietal cortical tissue and brains of R6/2 and YAC128 HD mouse models have been detected [70][71]. miR-132 is greatly enhanced in the brain [72][73], and its expression alters neuronal morphogenesis and sustains neurite outgrowth by inhibiting the GTPase-activating protein p250GAP [74]. An additional target of miR-132 is the acetylcholinesterase (ACHE), an enzyme involved in the degradation of the neurotransmitter acetylcholine at the synapse [75]. ACHE is strongly implicated in cognitive performances, and ACHE inhibitors are FDA-approved for the treatment of AD [75]. Thus, reduced miR-132 levels may negatively influence brain health, via the p250 GAP (reducing its inhibition) and ACHE deregulation.
4. miRNA Dysregulation and Mitochondrial Dysfunction in ALS
2.3. miRNA Dysregulation and Mitochondrial Dysfunction in ALS
ALS is a group of intricate multi-factorial neurodegenerative disorders. It is due to the selective deficiency of upper and lower motor neurons, thus leading to gradual skeletal muscle atrophy and death by respiration collapse after 2–5 years from the onset. ALS incidence is 2 per 100,000 persons per year. Mutations in ALS causing genes are related to the disorder in roughly 70% of the familial forms of ALS (FALS) and 15% of the sporadic forms of ALS (SALS) [76]. The familial ALS forms are only around 5% of cases. However, most cases are sporadic, which are phenotypically interchangeable from familial types, implying that there are shared pathways causing neuronal death [77].
Well-explored genetic causes of ALS are mutations or deletion of the SOD1 gene (
Figure 4). Recently, through advanced genomic screening tools, other genes related to ALS have been recognized, including TAR DNA-binding protein 43 (TARDBP), encoding TDP-43; fused in sarcoma (FUS) and chromosome 9 open reading frame 72 (C9ORF72). Notably, TDP-43 and FUS are RNA-binding proteins that function in mRNA and miRNA biogenesis [78][79]. Loffreda and collaborators [80] reported that miR-129-5p was upregulated in various models of SOD1 linked ALS and in peripheral blood mononuclear cells (PBMCs) of SALS patients. This study showed that upon an antisense oligonucleotide (ASO) inhibitor of miR-129-5p, ALS SOD1 (G93A) mice ameliorated the neuromuscular phenotype. In light of this, miR-129-5p may be a therapeutic target to treat ALS patients.
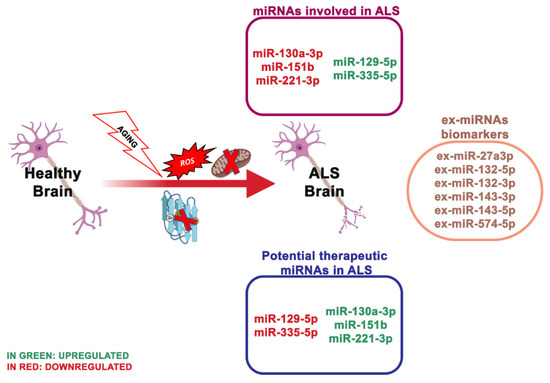
Figure 4.
Different mechanisms are responsible for the neuronal death occurring in ALS, including impaired metabolism, neuroinflammation, oxidative imbalance, mitochondrial dysfunction, glutamate excitotoxicity, growth factor defects and defective axonal transport [81][82]. Still, the pathogenic molecular mechanisms underlying ALS onset and progression are not entirely known.
To date, there is no available cure for this disorder. Increasing evidence reported that also the variation of RNA metabolism, including miRNA processing, is an essential pathogenetic component and a potential target for ALS.
Recently, the miRNA expression in ALS serum was analyzed and the assumption that changes of some miRNA involve the mitochondrial physiology of neuronal cells was tested [83]. The screening of numerous miRNAs in serum from patients and controls showed that miR335-5p was strongly decreased in ALS patients, and this was confirmed in an independent validation cohort.
MiR-335-5p targets 2544 genes [84], and its downregulation is involved in neuronal plasticity and memory processes in mice [85]. The downregulation of miR-335-5p induced mitophagy in SH-SY5Y cells and helped the concept that dysregulated miRNAs may participate in the pathogenesis of neuronal degeneration in ALS.
ALS lacks specific biomarkers, thus, the clinical diagnosis is problematic, with a high rate of misdiagnosis [86]. Notably, the serum levels of miR-1234-3p and miR-1825 resulted in being substantially reduced in ALS patients, and the miR-1825 decline was detected in both sporadic and familial ALS patients. At the same time, the miR-1234-3p reduction was limited to patients with sporadic ALS [87].
Plasma levels of miR-130a-3p, miR-151b and miR-221-3p resulted in also being reduced in sporadic ALS patients and positively associated with sporadic ALS progression, indicating that these miRNAs may be valuable even for monitoring the disorder progress [88]. A study of serum-derived ex-miRNA analyzed 10 ALS patients and 20 controls and revealed that ex-miR-27a-3p levels were considerably reduced in ALS patients. The researchers assumed that ex-miR-27a3p might represent a possible diagnostic biomarker of ALS [89] .
References
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47, doi:10.1016/j.mad.2016.01.002.
- Bonet-Costa, V.; Pomatto, L.C.-D.; Davies, K.J.A. The Proteasome and Oxidative Stress in Alzheimer’s Disease. Antioxid. Redox Signal. 2016, 25, 886–901, doi:10.1089/ars.2016.6802.
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1345–1367, doi:10.3233/JAD-170543.
- García-Escudero, V.; Martín-Maestro, P.; Perry, G.; Avila, J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell. Longev. 2013, 2013, 162152, doi:10.1155/2013/162152.
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6, doi:10.3390/biom6010006.
- Williamson, J.; Goldman, J.; Marder, K.S. Genetic aspects of Alzheimer disease. Neurologist 2009, 15, 80–86, doi:10.1097/NRL.0b013e318187e76b.
- Blass, J.P. Brain metabolism and brain disease: Is metabolic deficiency the proximate cause of Alzheimer dementia? J. Neurosci. Res. 2001, 66, 851–856, doi:10.1002/jnr.10087.
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492, doi:10.1016/j.mito.2012.10.011.
- Chen, F.; Zhao, Y.; Chen, H. MicroRNA-98 reduces amyloid β-protein production and improves oxidative stress and mitochondrial dysfunction through the Notch signaling pathway via HEY2 in Alzheimer’s disease mice. Int. J. Mol. Med. 2018, doi:10.3892/ijmm.2018.3957.
- Bian, S.; Sun, T. Functions of Noncoding RNAs in Neural Development and Neurological Diseases. Mol. Neurobiol. 2011, 44, 359–373, doi:10.1007/s12035-011-8211-3.
- Brai, E.; Alina Raio, N.; Alberi, L. Notch1 hallmarks fibrillary depositions in sporadic Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 64, doi:10.1186/s40478-016-0327-2.
- Li, Q.; Li, X.; Wang, L.; Zhang, Y.; Chen, L. miR-98-5p Acts as a Target for Alzheimer’s Disease by Regulating Aβ Production Through Modulating SNX6 Expression. J. Mol. Neurosci. 2016, 60, 413–420, doi:10.1007/s12031-016-0815-7.
- Hu, Y.-K.; Wang, X.; Li, L.; Du, Y.-H.; Ye, H.-T.; Li, C.-Y. MicroRNA-98 induces an Alzheimer’s disease-like disturbance by targeting insulin-like growth factor 1. Neurosci. Bull. 2013, 29, 745–751, doi:10.1007/s12264-013-1348-5.
- Zhou, Y.; Wang, Z.-F.; Li, W.; Hong, H.; Chen, J.; Tian, Y.; Liu, Z.-Y. Protective effects of microRNA-330 on amyloid β-protein production, oxidative stress, and mitochondrial dysfunction in Alzheimer’s disease by targeting VAV1 via the MAPK signaling pathway. J. Cell. Biochem. 2018, 119, 5437–5448, doi:10.1002/jcb.26700.
- Zhang, R.; Zhou, H.; Jiang, L.; Mao, Y.; Cui, X.; Xie, B.; Cui, D.; Wang, H.; Zhang, Q.; Xu, S. MiR-195 dependent roles of mitofusin2 in the mitochondrial dysfunction of hippocampal neurons in SAMP8 mice. Brain Res. 2016, 1652, 135–143, doi:10.1016/j.brainres.2016.09.047.
- Li, J.; Donath, S.; Li, Y.; Qin, D.; Prabhakar, B.S.; Li, P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010, 6, e1000795, doi:10.1371/journal.pgen.1000795.
- Ryan, M.M.; Guévremont, D.; Mockett, B.G.; Abraham, W.C.; Williams, J.M. Circulating Plasma microRNAs are Altered with Amyloidosis in a Mouse Model of Alzheimer’s Disease. JAD 2018, 66, 835–852, doi:10.3233/JAD-180385.
- Reddy, P.H.; Tonk, S.; Kumar, S.; Vijayan, M.; Kandimalla, R.; Kuruva, C.S.; Reddy, A.P. A critical evaluation of neuroprotective and neurodegenerative MicroRNAs in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1156–1165, doi:10.1016/j.bbrc.2016.08.067.
- Ravanidis, S.; Bougea, A.; Papagiannakis, N.; Maniati, M.; Koros, C.; Simitsi, A.; Bozi, M.; Pachi, I.; Stamelou, M.; Paraskevas, G.P.; et al. Circulating Brain‐Enriched MicroRNAs for Detection and Discrimination of Idiopathic and Genetic Parkinson’s Disease. Mov. Disord. 2020, 35, 457–467, doi:10.1002/mds.27928.
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS ONE 2015, 10, e0139233, doi:10.1371/journal.pone.0139233.
- Rani, A.; O’Shea, A.; Ianov, L.; Cohen, R.A.; Woods, A.J.; Foster, T.C. miRNA in Circulating Microvesicles as Biomarkers for Age-Related Cognitive Decline. Front. Aging Neurosci. 2017, 9, 323, doi:10.3389/fnagi.2017.00323.
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; Armengol, M.P.; Borràs, F.E.; Beyer, K. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl. Neurodegener. 2019, 8, 31, doi:10.1186/s40035-019-0169-5.
- Yang, T.T.; Liu, C.G.; Gao, S.C.; Zhang, Y.; Wang, P.C. The Serum Exosome Derived MicroRNA-135a, -193b, and -384 Were Potential Alzheimer’s Disease Biomarkers. Biomed. Environ. Sci. 2018, 31, 87–96, doi:10.3967/bes2018.011.
- Grasso, M.; Piscopo, P.; Crestini, A.; Confaloni, A.; Denti, M.A. Circulating microRNAs in Neurodegenerative Diseases. Exp. Suppl. 2015, 106, 151–169, doi:10.1007/978-3-0348-0955-9_7.
- Sanders, L.H.; Timothy Greenamyre, J. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 2013, 62, 111–120, doi:10.1016/j.freeradbiomed.2013.01.003.
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32, doi:10.1016/j.pneurobio.2013.04.004.
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-Synuclein Promotes Mitochondrial Deficit and Oxidative Stress. Am. J. Pathol. 2000, 157, 401–410, doi:10.1016/S0002-9440(10)64553-1.
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25, doi:10.1016/j.expneurol.2007.07.004.
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2–ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104, doi:10.1016/j.pharmthera.2015.11.003.
- Hoss, A.G.; Labadorf, A.; Beach, T.G.; Latourelle, J.C.; Myers, R.H. microRNA Profiles in Parkinson’s Disease Prefrontal Cortex. Front. Aging Neurosci. 2016, 8, 36, doi:10.3389/fnagi.2016.00036.
- Tan, L.; Yu, J.-T.; Tan, L. Causes and Consequences of MicroRNA Dysregulation in Neurodegenerative Diseases. Mol. Neurobiol. 2015, 51, 1249–1262, doi:10.1007/s12035-014-8803-9.
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA Feedback Circuit in Midbrain Dopamine Neurons. Science 2007, 317, 1220–1224, doi:10.1126/science.1140481.
- Junn, E.; Lee, K.-W.; Jeong, B.S.; Chan, T.W.; Im, J.-Y.; Mouradian, M.M. Repression of -synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057, doi:10.1073/pnas.0906277106.
- Doxakis, E. Post-transcriptional Regulation of α-Synuclein Expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734, doi:10.1074/jbc.M109.086827.
- Itoh, N.; Ohta, H. Roles of FGF20 in dopaminergic neurons and Parkinson’s disease. Front. Mol. Neurosci. 2013, 6, 15, doi:10.3389/fnmol.2013.00015.
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053, doi:10.18632/oncotarget.6158.
- Lin, W.; Metzakopian, E.; Mavromatakis, Y.E.; Gao, N.; Balaskas, N.; Sasaki, H.; Briscoe, J.; Whitsett, J.A.; Goulding, M.; Kaestner, K.H.; et al. Foxa1 and Foxa2 function both upstream of and cooperatively with Lmx1a and Lmx1b in a feedforward loop promoting mesodiencephalic dopaminergic neuron development. Dev. Biol. 2009, 333, 386–396, doi:10.1016/j.ydbio.2009.07.006.
- Kittappa, R.; Chang, W.W.; Awatramani, R.B.; McKay, R.D.G. The foxa2 Gene Controls the Birth and Spontaneous Degeneration of Dopamine Neurons in Old Age. PLoS Biol. 2007, 5, e325, doi:10.1371/journal.pbio.0050325.
- Parker, W.D.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218, doi:10.1016/j.brainres.2007.10.061.
- Wang, H.; Ye, Y.; Zhu, Z.; Mo, L.; Lin, C.; Wang, Q.; Wang, H.; Gong, X.; He, X.; Lu, G.; et al. MiR-124 Regulates Apoptosis and Autophagy Process in MPTP Model of Parkinson’s Disease by Targeting to Bim. Brain Pathol. 2016, 26, 167–176, doi:10.1111/bpa.12267.
- Hayashi, T.; Ishimori, C.; Takahashi-Niki, K.; Taira, T.; Kim, Y.; Maita, H.; Maita, C.; Ariga, H.; Iguchi-Ariga, S.M.M. DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 2009, 390, 667–672, doi:10.1016/j.bbrc.2009.10.025.
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.C.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746, doi:10.1093/hmg/ddq288.
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B.; Wang, L.; Wu, L.; Li, W.; Ding, J.; et al. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714, doi:10.1016/j.neurobiolaging.2013.09.027.
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078, doi:10.1093/hmg/ddr210.
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020, doi:10.1212/WNL.0b013e3181ff9685.
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944, doi:10.1093/hmg/dds003.
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620, doi:10.1093/hmg/dds470.
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-α regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 451–461, doi:10.1016/j.bbadis.2014.11.019.
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression. J. Biol. Chem. 2016, 291, 6483–6493, doi:10.1074/jbc.M115.691352.
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1 , A Potential Therapeutic Target for Early Intervention in Parkinson’s Disease. Sci. Transl. Med. 2010, 2, 52ra73, doi:10.1126/scitranslmed.3001059.
- Wang, Y.; Yang, Z.; Le, W. Tiny But Mighty: Promising Roles of MicroRNAs in the Diagnosis and Treatment of Parkinson’s Disease. Neurosci. Bull. 2017, 33, 543–551, doi:10.1007/s12264-017-0160-z.
- Valdés, P.; Schneider, B.L. Gene Therapy: A Promising Approach for Neuroprotection in Parkinson’s Disease? Front. Neuroanat. 2016, 10, 123, doi:10.3389/fnana.2016.00123.
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228, doi:10.1016/S0140-6736(07)60111-1.
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, doi:10.1101/cshperspect.a007476.
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403, doi:10.1038/ng0893-398.
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342, doi:10.3389/fnins.2018.00342.
- Gutekunst, C.-A.; Li, S.-H.; Yi, H.; Ferrante, R.J.; Li, X.-J.; Hersch, S.M. The Cellular and Subcellular Localization of Huntingtin-Associated Protein 1 (HAP1): Comparison with Huntingtin in Rat and Human. J. Neurosci. 1998, 18, 7674–7686, doi:10.1523/JNEUROSCI.18-19-07674.1998.
- Choo, Y.S. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420, doi:10.1093/hmg/ddh162.
- Orr, A.L.; Li, S.; Wang, C.-E.; Li, H.; Wang, J.; Rong, J.; Xu, X.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.-J. N-Terminal Mutant Huntingtin Associates with Mitochondria and Impairs Mitochondrial Trafficking. J. Neurosci. 2008, 28, 2783–2792, doi:10.1523/JNEUROSCI.0106-08.2008.
- Puigserver, P.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocr. Rev. 2003, 24, 78–90, doi:10.1210/er.2002-0012.
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69, doi:10.1016/j.cell.2006.09.015.
- Stanek, L.M.; Bu, J.; Shihabuddin, L.S. Astrocyte transduction is required for rescue of behavioral phenotypes in the YAC128 mouse model with AAV-RNAi mediated HTT lowering therapeutics. Neurobiol. Dis. 2019, 129, 29–37, doi:10.1016/j.nbd.2019.04.015.
- Bradford, J.; Shin, J.-Y.; Roberts, M.; Wang, C.-E.; Li, X.-J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. PNAS 2009, 106, 22480–22485, doi:10.1073/pnas.0911503106.
- Skotte, N.H.; Andersen, J.V.; Santos, A.; Aldana, B.I.; Willert, C.W.; Nørremølle, A.; Waagepetersen, H.S.; Nielsen, M.L. Integrative Characterization of the R6/2 Mouse Model of Huntington’s Disease Reveals Dysfunctional Astrocyte Metabolism. Cell Rep. 2018, 23, 2211–2224, doi:10.1016/j.celrep.2018.04.052.
- Cheng, P.-H.; Li, C.-L.; Chang, Y.-F.; Tsai, S.-J.; Lai, Y.-Y.; Chan, A.W.S.; Chen, C.-M.; Yang, S.-H. miR-196a Ameliorates Phenotypes of Huntington Disease in Cell, Transgenic Mouse, and Induced Pluripotent Stem Cell Models. Am. J. Hum. Genet. 2013, 93, 306–312, doi:10.1016/j.ajhg.2013.05.025.
- Gaughwin, P.M.; Ciesla, M.; Lahiri, N.; Tabrizi, S.J.; Brundin, P.; Björkqvist, M. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 2011, 20, 2225–2237, doi:10.1093/hmg/ddr111.
- Langfelder, P.; Gao, F.; Wang, N.; Howland, D.; Kwak, S.; Vogt, T.F.; Aaronson, J.S.; Rosinski, J.; Coppola, G.; Horvath, S.; et al. MicroRNA signatures of endogenous Huntingtin CAG repeat expansion in mice. PLoS ONE 2018, 13, e0190550, doi:10.1371/journal.pone.0190550.
- Li, X.; Standley, C.; Sapp, E.; Valencia, A.; Qin, Z.-H.; Kegel, K.B.; Yoder, J.; Comer-Tierney, L.A.; Esteves, M.; Chase, K.; et al. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol. Cell. Biol. 2009, 29, 6106–6116, doi:10.1128/MCB.00420-09.
- Hoss, A.G.; Labadorf, A.; Latourelle, J.C.; Kartha, V.K.; Hadzi, T.C.; Gusella, J.F.; MacDonald, M.E.; Chen, J.-F.; Akbarian, S.; Weng, Z.; et al. miR-10b-5p expression in Huntington’s disease brain relates to age of onset and the extent of striatal involvement. BMC Med. Genom. 2015, 8, 10, doi:10.1186/s12920-015-0083-3.
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008, 29, 438–445, doi:10.1016/j.nbd.2007.11.001.
- Lee, S.-T.; Chu, K.; Im, W.-S.; Yoon, H.-J.; Im, J.-Y.; Park, J.-E.; Park, K.-H.; Jung, K.-H.; Lee, S.K.; Kim, M.; et al. Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 2011, 227, 172–179, doi:10.1016/j.expneurol.2010.10.012.
- Lagos-Quintana, M. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858, doi:10.1126/science.1064921.
- Miska, E.A.; Alvarez-Saavedra, E.; Townsend, M.; Yoshii, A.; Sestan, N.; Rakic, P.; Constantine-Paton, M.; Horvitz, H.R. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004, 5, R68, doi:10.1186/gb-2004-5-9-r68.
- Vo, N.; Klein, M.E.; Varlamova, O.; Keller, D.M.; Yamamoto, T.; Goodman, R.H.; Impey, S. From The Cover: A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 16426–16431, doi:10.1073/pnas.0508448102.
- McGleenon, B.M.; Dynan, K.B.; Passmore, A.P. Acetylcholinesterase inhibitors in Alzheimer’s disease: Acetylcholinesterase inhibitors in Alzheimer’s disease. Br. J. Clin. Pharmacol. 2001, 48, 471–480, doi:10.1046/j.1365-2125.1999.00026.x.
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102, doi:10.1016/S1474-4422(17)30401-5.
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 679–684, doi:10.1016/j.bbadis.2014.08.010.
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352, doi:10.1073/pnas.1112427109.
- Buratti, E.; De Conti, L.; Stuani, C.; Romano, M.; Baralle, M.; Baralle, F. Nuclear factor TDP-43 can affect selected microRNA levels: TDP-43 and miRNA regulation. FEBS J. 2010, 277, 2268–2281, doi:10.1111/j.1742-4658.2010.07643.x.
- Loffreda, A.; Nizzardo, M.; Arosio, A.; Ruepp, M.-D.; Calogero, R.A.; Volinia, S.; Galasso, M.; Bendotti, C.; Ferrarese, C.; Lunetta, C.; et al. miR-129-5p: A key factor and therapeutic target in amyotrophic lateral sclerosis. Prog. Neurobiol. 2020, 190, 101803, doi:10.1016/j.pneurobio.2020.101803.
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98, doi:10.1093/bmb/ldw026.
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611, doi:10.1016/j.bbrc.2006.10.093.
- De Luna, N.; Turon-Sans, J.; Cortes-Vicente, E.; Carrasco-Rozas, A.; Illán-Gala, I.; Dols-Icardo, O.; Clarimón, J.; Lleó, A.; Gallardo, E.; Illa, I.; et al. Downregulation of miR-335-5P in Amyotrophic Lateral Sclerosis Can Contribute to Neuronal Mitochondrial Dysfunction and Apoptosis. Sci. Rep. 2020, 10, 4308, doi:10.1038/s41598-020-61246-1.
- Hamzeiy, H.; Suluyayla, R.; Brinkrolf, C.; Janowski, S.J.; Hofestädt, R.; Allmer, J. Visualization and Analysis of miRNAs Implicated in Amyotrophic Lateral Sclerosis Within Gene Regulatory Pathways. Stud. Health Technol. Inform. 2018, 253, 183–187.
- Capitano, F.; Camon, J.; Licursi, V.; Ferretti, V.; Maggi, L.; Scianni, M.; Del Vecchio, G.; Rinaldi, A.; Mannironi, C.; Limatola, C.; et al. MicroRNA-335-5p modulates spatial memory and hippocampal synaptic plasticity. Neurobiol. Learn. Mem. 2017, 139, 63–68, doi:10.1016/j.nlm.2016.12.019.
- Wang, L.; Zhang, L. Circulating Exosomal miRNA as Diagnostic Biomarkers of Neurodegenerative Diseases. Front. Mol. Neurosci. 2020, 13, 53, doi:10.3389/fnmol.2020.00053.
- Freischmidt, A.; Müller, K.; Zondler, L.; Weydt, P.; Mayer, B.; von Arnim, C.A.F.; Hübers, A.; Dorst, J.; Otto, M.; Holzmann, K.; et al. Serum microRNAs in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 2660.e15–2660.e20, doi:10.1016/j.neurobiolaging.2015.06.003.
- Liguori, M.; Nuzziello, N.; Introna, A.; Consiglio, A.; Licciulli, F.; D’Errico, E.; Scarafino, A.; Distaso, E.; Simone, I.L. Dysregulation of MicroRNAs and Target Genes Networks in Peripheral Blood of Patients With Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 11, 288, doi:10.3389/fnmol.2018.00288.
- Xu, Z.; Henderson, R.D.; David, M.; McCombe, P.A. Neurofilaments as Biomarkers for Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0164625, doi:10.1371/journal.pone.0164625.
- Freischmidt, A.; Müller, K.; Ludolph, A.C.; Weishaupt, J.H. Systemic dysregulation of TDP-43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2013, 1, 42, doi:10.1186/2051-5960-1-42.