The retina is an exquisite target for defects of oxidative phosphorylation (OXPHOS) associated with mitochondrial impairment. Retinal involvement occurs in two ways, retinal dystrophy (retinitis pigmentosa) and subacute or chronic optic atrophy, which are the most common clinical entities. Both can present as isolated or virtually exclusive conditions, or as part of more complex, frequently multisystem syndromes. In most cases, mutations of mtDNA have been found in association with mitochondrial retinopathy. The main genetic abnormalities of mtDNA include mutations associated with neurogenic muscle weakness, ataxia and retinitis pigmentosa (NARP) sometimes with earlier onset and increased severity (maternally inherited Leigh syndrome, MILS), single large-scale deletions determining Kearns–Sayre syndrome (KSS, of which retinal dystrophy is a cardinal symptom), and mutations, particularly in mtDNA-encoded ND genes, associated with Leber hereditary optic neuropathy (LHON). However, mutations in nuclear genes can also cause mitochondrial retinopathy, including autosomal recessive phenocopies of LHON, and slowly progressive optic atrophy caused by dominant or, more rarely, recessive, mutations in the fusion/mitochondrial shaping protein OPA1, encoded by a nuclear gene on chromosome 3q29.
- retina
- mitochondrial disorders
- mitochondrial DNA
- retinitis pigmentosa
1. Mitochondrial Bioenergetics
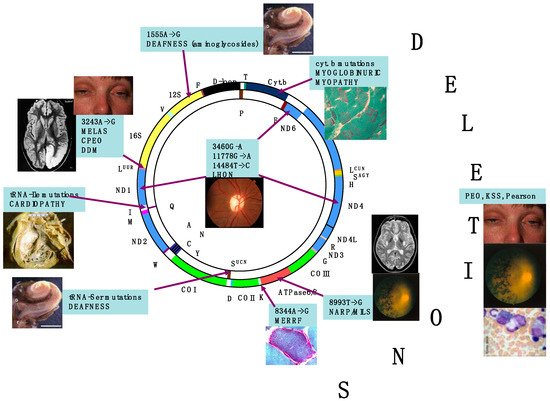
2. Retina Is a Preferential Target of Mitochondrial Dysfunction and Diseases
3. Functional Anatomy of the Retina
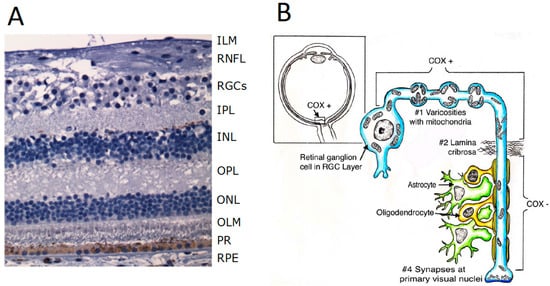
4. Physiology of the Photoreceptors
-
The photoreceptors—rods and cones—which transmit signals to the outer plexiform layer, where they synapse with bipolar cells and horizontal cells.
-
The horizontal cells, which transmit signals horizontally in the outer plexiform layer from the rods and cones to bipolar cells.
-
The bipolar cells, which transmit signals vertically from the rods, cones, and horizontal cells to the inner plexiform layer, where they synapse with ganglion cells and amacrine cells.
-
The amacrine cells, which transmit signals in two directions, either directly from bipolar cells to RGCs or horizontally within the inner plexiform layer from axons of the bipolar cells to dendrites of the ganglion cells or to other amacrine cells.
-
The ganglion cells, or RGCs, which transmit output signals from the retina through the optic nerve into the brain serving both the visual and non-visual pathways (mRGCs). The first leads to formed vision, whereas the second is instrumental to photoentrain circadian rhythms.
References
- Wallace, D.C. Mitochondria, bioenergetics, and the epigenome in eukaryotic and human evolution. Cold Spring Harb. Symp. Quant. Biol. 2009, 74, 383–393.
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407.
- Dzbek, J.; Korzeniewski, B. Control over the contribution of the mitochondrial membrane potential (ΔΨ) and proton gradient (ΔpH) to the protonmotive force (Δp). J. Biol. Chem. 2008, 283, 33232–33239.
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation (1966). Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1507–1538.
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714.
- Montava-Garriga, L.; Ganley, I.G. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 2020, 432, 206–230.
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genom. Hum. Genet. 2010, 11, 25–44.
- Wallace, D.C. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu. Rev. Biochem. 2007, 76, 781–821.
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080.
- Zeviani, M.; Di Donato, S. Mitochondrial disorders. Brain 2004, 127, 2153–2172.
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444.
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory−chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668.
- Schon, E.A.; DiMauro, S. Mitochondrial mutations: Genotype to phenotype. Novartis Found. Symp. 2007, 287, 214–225.
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010, 9, 829–840.
- Carelli, V.; Barboni, P.; Sadun, A.A. Mitochondrial ophthalmology. In Mitochondrial Medicine; DiMauro, S., Hirano, M., Schon, E., Eds.; Informa Healthcare: Oxford, UK, 2006.
- Miller, N.R.; Newman, N.J.; Biousse, V.; Kerrison, J.B. (Eds.) Walsh & Hoyt’s Clinical Neuro-Ophthalmology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005; ISBN 0-7817-4814-3.
- La Morgia, C.; Carelli, V.; Sadun, A.A. Retina and melanopsin neurons. Handb. Clin. Neurol. 2021, 179, 315–329.
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881.
- Steinberg, R.H.; Linsenmeier, R.A.; Griff, E.R. Three light-evoked responses of the retinal pigment epithelium. Vis. Res. 1983, 23, 1315–1323.