Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by vaiman Vaiman and Version 2 by Vicky Zhou.
Oxidative stress (OS) plays a pivotal role in placental development; however, abnormal loads in oxidative stress molecules may overwhelm the placental defense mechanisms and cause pathological situations. The environment in which the mother evolves triggers an exposure of the placental tissue to chemical, physical, and biological agents of OS, with potential pathological consequences.
- environmental pollution
- placenta
- heavy metals
- P38MAPK
- DNA methylation
- placental diseases
- hypertensive disorders of pregnancy
1. Introduction
The placenta is an extraembryonic annex that develops as a major interface between the mother and the fetus in mammals. The placenta allows the exchanges of nutrients, gases, the discard of metabolic waste, and is operative for the synthesis of gestation-essential hormones responsible for placental growth, angiogenesis. The placenta is also essential for the immune tolerance of the mother towards the hemi-allogenic fetus and the dampening of the maternal immunological reactions throughout pregnancy.
One accompanying issue of placentation is the ubiquity of oxidative stress (OS). In a broad sense, OS is directly linked to the generation of chemicals that have a strong chemical affinity for biomolecules (proteins, lipids, DNA). When an excess of these reactive molecules is generated, it gives rise to OS. Despite being necessary for proper placental development, OS can have deleterious effects if an extreme dysregulation occurs. Elevated OS can induce complications in pregnancy progression, leading to long-term effects on both the mother and the fetus.
2. Mechanisms of Oxidative Stress Induction by Environmental Pollutants
Environmental pollutants are produced mainly as a consequence of human activities, but also by other living organisms or natural causes, and are ubiquitously distributed in air, soil, water, food, plastics, etc. Therefore, all living organisms, including humans, are exposed to a variety of pollutants throughout their life. Epidemiological evidence shows that many of these pollutants can adversely impact pregnancy. Pending on the nature of the pollutant, the health of the mother, the fetus, or both, can be seriously affected. The toxic effects of pollutants can lead to embryonic mortality, spontaneous abortion, IUGR, low birth weight, neurophysiological pathologies, etc. [1][2][3][4][24,25,26,27]. The mechanisms of action of these pollutants are diverse, but in many cases, induction of OS has been identified as a common mechanism of action [3][5][6][26,28,29]. In this section, we will thus focus on those environmental pollutants which have been found in the placenta or conceptus and are known to act through the induction of OS. These pollutants belong to several categories, heavy metals, air pollutants (fine particles, tobacco smoke), pollutants of biological origin (toxins), industrial molecules used in the production of plastics, and domestic goods (phenols and parabens). Here we will focus on the cases where some degree of mechanistic information has been gained through research. The enhancement of ROS production and the induction of cellular damage via OS is a common finding in the studies analyzing the mode of action of environmental pollutants. In most cases, however, the precise mechanisms underlying the increased production of ROS by environmental pollutants remain to be discovered and analyzed. Nevertheless, several studies distinguish between direct and indirect mechanisms. Direct mechanisms involve the production of ROS as a result of the chemical reaction of the pollutant with other molecules present inside the cell. Indirect effects involve the capacity of some pollutants to inhibit ROS scavenging antioxidant enzymes, but also their capacity to inhibit the electron transport chain (ETC) of the mitochondria with the subsequent increase in superoxide production and decreased ATP generation.2.1. Exposure to Environmental Metals
The induction of ROS production has been studied with some detail in the case of heavy metals [7][30]. These exposures are detectable in the maternal blood. This has recently been studied for 41 metals and metalloids in the Shanxi province of China [8][31]. OS is detectable on DNA through 8-OHdG labeling; 8-Oxo-7,8-dihydroguanine (8-OHdG) is widely used as a predominant biomarker for OS, with the advantage of being relatively stable and easy to study by validated ELISA tests [9][32]. The Singh study attempted to connect the metal/metalloid exposure to OS to the risk of spontaneous preterm birth (74 cases, 74 controls). The study revealed that exposure to lutetium, erbium, europium, praseodymium, and iron indeed increased the marks of OS in the maternal DNA. This suggests that placental alterations similar to those of the maternal DNA are plausible, although they have not been directly studied by the authors.
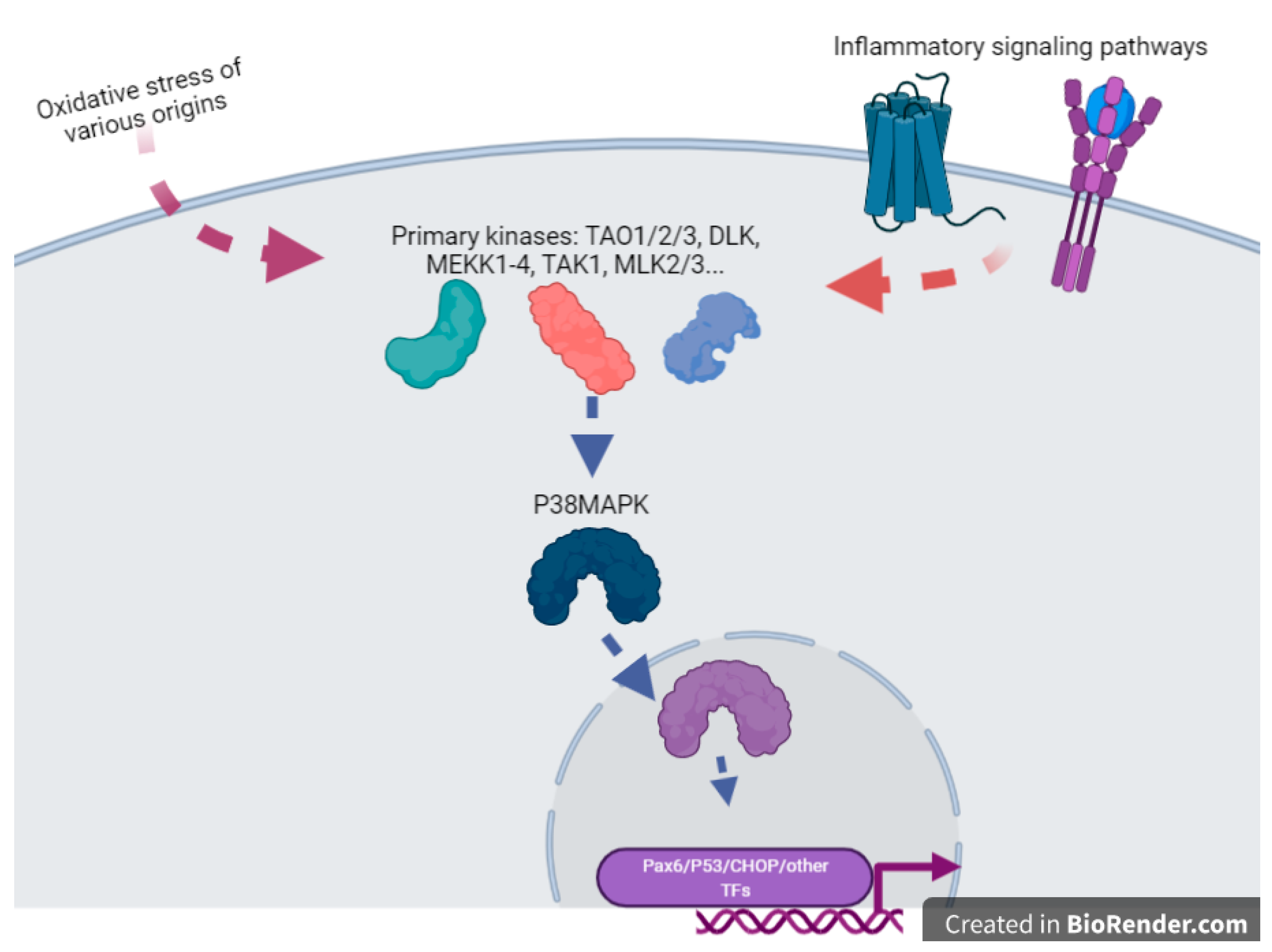 Figure 12. A cartoon emphasizing the pivotal role of P38MAPK(MAPK11) in the sensing of OS and the direct or indirect activation of various transcription factors. This is an oversimplification since this factor, once inside the nucleus, can also modulate chromatin structure through action upon structural components of the chromatins such as HMGN1 or Histone H3. This drawing is a simplification from [14][89].
Figure 12. A cartoon emphasizing the pivotal role of P38MAPK(MAPK11) in the sensing of OS and the direct or indirect activation of various transcription factors. This is an oversimplification since this factor, once inside the nucleus, can also modulate chromatin structure through action upon structural components of the chromatins such as HMGN1 or Histone H3. This drawing is a simplification from [14][89].
2.2. Exposure to Tobacco
Cigarette smoke extract exposure in vivo by injection in the uterine sacs of mice induce specific protein damages, protein nitrotyrosylation [10][85], especially with the activation of p38MAPK (aka MAPK11), a major actor of inflammatory processes, such as TLR signaling [11][86]. In these reactions, the activity of p38MAPK seems crucial (Figure 12), and the activity of this enzyme appeared induced by OS leading to inflammation in various cell types, and various studies indicate that this molecule can be relatively specifically targeted to alleviate defects induced by OS, for instance with specific inhibitors (SB203580) or statins, molecules that are well-known for their anti-cholesterol effects. P38MAPK could be important for the regulation of senescence, and all the senescence processes are pivotal in gestation since they are the monitors of placental ageing. Abnormal regulation of placental ageing has been involved in various placental pathologies, such as PE or the premature rupture of fetal membranes, leading to preterm birth. Tobacco smoke extracts accelerate the senescence, and there are concrete elements linking in a cascade: tobacco smoke ➔ oxidative stress ➔ p38MAPK activation ➔ senescence ➔ placental disease; these mechanisms may be counterbalanced by simvastatin and rosuvastatin treatment [12][87]. This implication of the p38MAPK cascade was also evinced in the mouse model [10][85]. Three kinase signaling pathways co-exist in eukaryotic cells (p38MAPK, ERK, and JNK (aka MAPK8)). The two first kinases can activate the COX-2 (PTGS2) prostaglandin inflammatory pathway (leading to PGE2 synthesis), while the JNK pathway tends to allow direct biogenesis of PGE2 without activating PTGS2. Similar to p38, cigarette smoke extract may also act through ERK1/2 activation [13][88]. This suggests that activation of ROS through the ERK1/2-EGR1 axis may also potentially contribute to the production of PlGF since EGR1 binds to the PlGF promoter. In this case, since PlGF is a favorable cofactor in pregnancy, this pathway could represent a normal contribution of ROS to increased angiogenesis in the early placenta, after the spiral arteries trophoblast plugs are expelled, at the dawn of the second trimester of pregnancy.
Figure 12. A cartoon emphasizing the pivotal role of P38MAPK(MAPK11) in the sensing of OS and the direct or indirect activation of various transcription factors. This is an oversimplification since this factor, once inside the nucleus, can also modulate chromatin structure through action upon structural components of the chromatins such as HMGN1 or Histone H3. This drawing is a simplification from [14][89].
2.3. Exposure to Airborne Particulate Matter
Airborne particulate matter (PM) is an important pollutant of the urban atmosphere and has been linked to OS and inflammation [15][105]. The particles are classified according to their diameter as PM2.5 or PM10 for example (<2.5 or 10 µM, respectively). It has been shown that air pollution particles may translocate through the STB membrane and cross the placental barrier [16][106]. PM from combustion sources contains a number of constituents that generate ROS by a variety of reactions [17][107]. A population study published in 2016 indicates clearly that this OS is combined with a proportion of nitrosative stress, a type of stress promoted by the generation of peroxynitrite molecules, ultimately leading to the covalent coupling of 3-nitrotyrosine to proteins causing their altered function [18][108]. This specific mark has recently been shown to be a premise of neurogenerative diseases, present before the onset of symptoms [19][109]. In the paper by Saenen and coworkers [18][108], 330 mother newborn couples revealed an increase by 35% of the level of nitrotyrosinated proteins, in association with an increase of 29% or 39% of the PM2.5, according to definite exposure windows (first and second trimester, respectively, while the third was not significant). In addition, it has now been shown that exposure to PM leads to locus-specific DNA methylation alterations [20][110]. One of the clearly identified loci was in the vicinity of the gene ADORA2B. Interestingly, this gene was associated to hyperglycemia and OS in several contexts, in particular gestational diabetes mellitus [21][22][111,112].
Several publications relate air pollution particle exposure to the oxidation of DNA [23][113]. In nuclear and mitochondrial DNA (mtDNA), the free radicals induced oxidative lesions. An examination of the effects of PM (2.5 and 10 μm diameter) exposure during pregnancy revealed an associated increase of mitochondrial 8-OHdG in maternal and cord blood of newborns. Thus, air pollution exposure in early life has a role in increasing systemic oxidative stress and DNA damage, at the level of the mitochondria, both in the mother and foetus [24][114]. Another study found an association between PM2.5 μm exposure during pregnancy and placental mtDNA methylation (especially in the MT-RNR1 region). According to the authors, this increased mtDNA methylation could reflect signs of mitophagy and mitochondrial death and result from PM induced oxidative stress [25][115].
2.4. Exposure to Vexing Biomolecules and Riling Toxins
Deoxynivalenol (DON) belongs to the type B group of the trichothecenes family, which is composed of sesquiterpenoid metabolites produced by Fusarium and other fungi in crops used for food and feed production [26][116]. Epidemiological studies have documented that DON affects animal and human health by causing various toxicities [27][117]. DON can transport across the placental barrier [28][118], and in pregnant mice, a relatively low-dose maternal DON exposure can result in developmental toxicities for embryos [29][119]. DON induces excessive accumulation of ROS, which leads to structural and functional damages of the placenta, causing adverse pregnancy outcomes. An experimental study using pregnant mice and BeWo cells has shown that DON exposure activates the Nrf2/HO-1 pathway and the expressions while its downstream antioxidant enzymes are increased to protect the placenta against oxidative stress [30][31][120,121]. However, with a longer time and higher dose exposure, the antioxidant capacity of cells reaches its limit. According to the authors, the reason of this “threshold effect” could be a consequence of the continuous release of iron by the HO-1 activity. This iron would then be involved in deleterious reactions that compete with iron reutilization, sequestration pathways, and aggravate oxidative stress [32][122].
The T-2 toxin is another major Fusarium mycotoxin contaminating crops [33][123]. In pregnant animals, the T-2 toxin induces placental lesions, embryotoxicity and abnormal development of offspring [33][34][123,124]. It has been shown that the T-2 toxin induces OS in placenta; however, its precise mode of action remains to be explored [35][125].
Microcystin-LR (MCLR) is another toxin able to impact human and animal reproduction [36][126]. MCLR is an environmental pollutant released by cyanobacteria in freshwater [37][127]. MCLR is actively absorbed by animals, fish, and birds from intoxicated water and thus enter the food chain. Humans are also exposed to microcystins, for instance, when they perform leisure activities in contaminated water. Pregnant mice intraperitoneally injected with MCLR (5 or 20 μg/kg) from gestational day (GD) 13 to GD17 present with reduced placenta and offspring weight. Expression of genes encoding placental growth factors Vegfα and Pgf, and transport pumps Glut1 and Pcft are in this case dampened in the placentas. Moreover, significant increases in MDA revealed the occurrence of OS caused by MCLR, which was also verified by a remarkable decrease in the glutathione levels, total antioxidant capacity (T-AOC), as well as the activity of antioxidant enzymes [38][128]. Moreover, MCLR activates the endoplasmic reticulum (ER) stress pathway in the placentas.
2.5. Exposure to Organometallic Molecules and Endocrine Disrupters
Tributyltin (TBT) is a persistent organotin pollutant widely used as agricultural and wood biocides for more than 40 years. Studies in mice have shown that it adversely impacts pregnancy by inducing developmental disorders of the placenta, including dysregulation of key molecules, impairing proliferation, inducing apoptosis, and oxidative stress. TBT administration increased levels of MDA and H2O2 and decreased activities of catalase and SOD [39][131]. Tributyltin also plays the role of an endocrine disrupter, as other organotins are supposed to act. A study in rats showed that organotin-contaminated sea food triggers abnormal histology of the placenta, accompanied by an excess of OS [40][132]. The effects of endocrine disrupters may be equivocal, as one study on the rat model indicates that exposure to genistein (a natural estrogeno-mimetic abundant in soy) modifies the OS status of the placenta [41][133]. Rats force-fed with genistein presented an increase of antioxidant levels (SOD, GSH, CAT) at gestational days 18 and 20 in the circulating blood, while an opposite effect was apparent in the placenta and amniotic fluid. According to the authors, this profile marks a positive effect of genistein attenuating OS in the placenta.
2.6. Plastic Modifiers: Phenols, Bisphenols and Parabens
Synthetic phenols constitute a family of chemicals supposed to have endocrine disruption properties. They are widely used to produce polycarbonate and epoxy resins, as well as ultraviolet filters, biocides (such as insecticides), antimicrobial agents for the fabrication of personal care products and plastics, and have become ubiquitous environmental contaminants. The general population is widely exposed to these molecules which are readily detected in placental tissues [15][42][43][44][105,134,135,136]. Studies in humans and animal models suggest that exposure to these compounds may be related to several adverse health outcomes, including pregnancy complications [45][46][47][48][49][50][137,138,139,140,141,142].
Imprinted genes are known to be targeted by these molecules [51][143], suggesting possible genomic (epigenetic effects through methylation alteration, as recently explored for nine molecules (bisphenolA -BPA-, benzophenone3, triclosan, 2.4 and 2.5 dichlorophenol, butyl- ethyl, methyl, and propylparaben). Altered methylation in the placentas was observed for 46 DMRs following exposition to triclosan (37 DMRs), confirming alterations nearby imprinted genes, nevertheless without specific alterations nearby genes directly involved in OS metabolism [52][144]. Consistently, Basak and coworkers showed in the trophoblast model HTR8/SV-neo that BPA exposure (1 nM) decreased promoter methylation of genes involved in metabolic and OS, such as GSR, PRDX2, GPX7, and GPX3, among others [53][145].
Examination of the mode of action of phthalates and parabens in the liver has also shown that they induce OS through the inhibition of the enzymatic activities of SOD, CAT, and GPX [54][55][146,147].
Tri-ortho-cresyl phosphate (TOCP), used as plasticizers, plastic softener, and flame-retardant, was reported to cause reproductive toxicity in mammals. In a recent study, dams were orally administered different doses of TOCP to explore its effects on placental development [56][148]. TOCP exposure significantly reduced numbers of the implanted embryo and adversely affected placental anthropometry. In addition to inducing apoptosis and autophagy, TOCP exposure increased the production of H2O2 and MDA. The induction of OS could be explained by an observed marked reduction in the activities of the catalase and SOD enzymes.
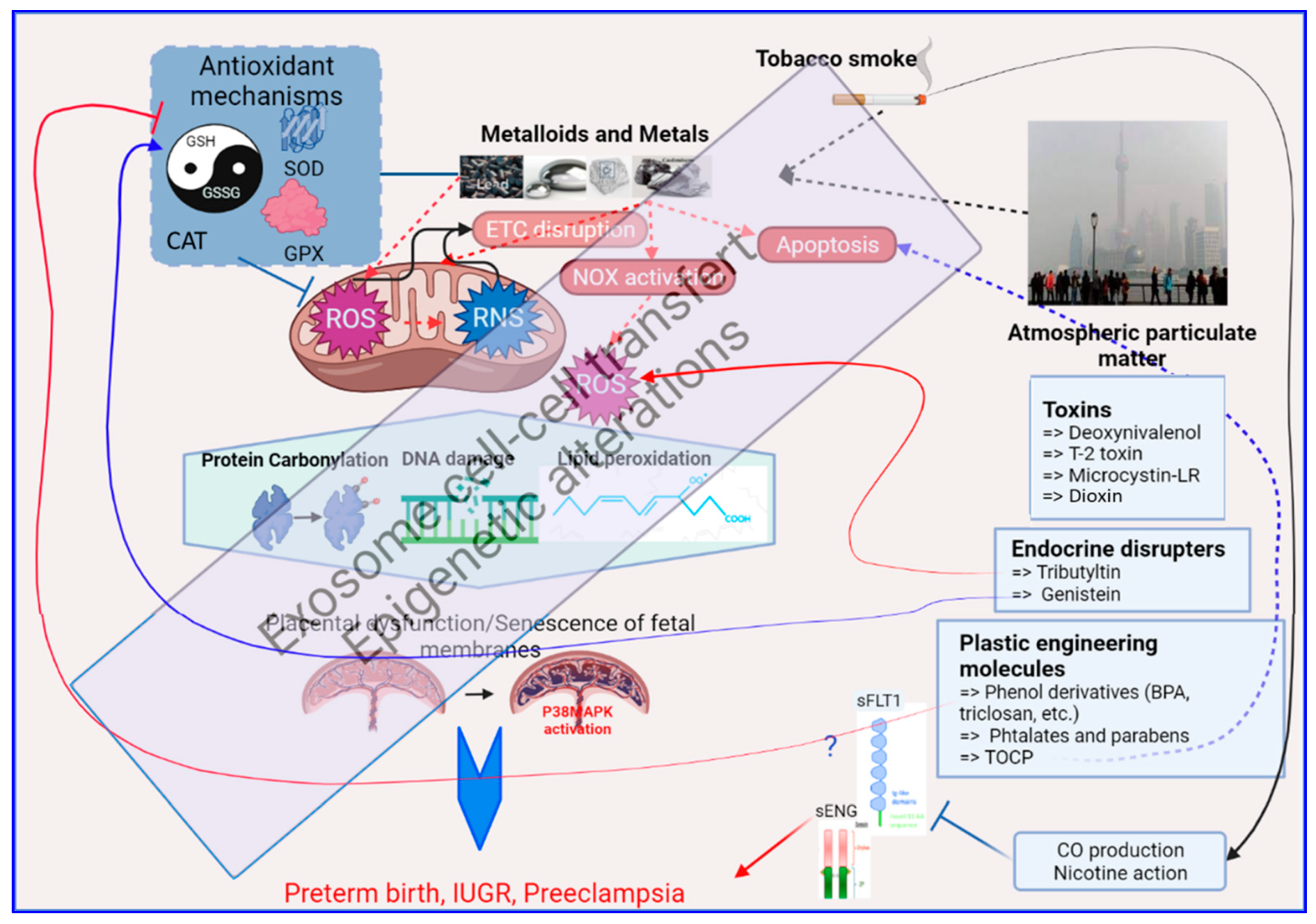 Figure 23. A brief graphic summary of the mechanistic impacts of environmental exposure through oxidative stress alterations in the human placenta, as detailed in the text of this review. At the center of this graph is the mitochondria which is the major producer of oxidative stress (ROS = Reactive Oxygen Species, and RNS = Reactive Nitrogen Species). The red dashed arrows relate to the generation of oxidative stress; the box on the upper left summarizes the antioxidant mechanisms that will fight against oxidative stress (the balance GSH/GSSG, the Superoxide Dismutases, the Glutathione peroxidases, the Catalase). The effect of OS on biomolecules appears on proteins, DNA and lipids as shown in the hexagon in the middle of the figure, with specific chemical modifications. Below, the placenta on which this review is focused is presented, part of the oxidative stress response being triggered in this organ by the activation of the P38/MAPK pathway. In black are the sources of oxidative stress in the human placenta that are described in this paper (Metalloids and Metals, Tobacco smoke and Atmospheric particulate matter). In Blue boxes at the right part of the figure are presented specific chemicals that are part of the placental regulation of oxidative stress, acting either on ROS production or modulation of ROS detoxification that is further detailed in the text. Other abbreviations: ETC = Electron Transport Chain, NOX = Nitric Oxidases, sFLT1 = Soluble FMS-Like Tyrosine kinase 1, sENG = soluble Endoglin, CO = Carbon Monoxide, TOCP = Tri-ortho-cresyl phosphate.
Figure 23. A brief graphic summary of the mechanistic impacts of environmental exposure through oxidative stress alterations in the human placenta, as detailed in the text of this review. At the center of this graph is the mitochondria which is the major producer of oxidative stress (ROS = Reactive Oxygen Species, and RNS = Reactive Nitrogen Species). The red dashed arrows relate to the generation of oxidative stress; the box on the upper left summarizes the antioxidant mechanisms that will fight against oxidative stress (the balance GSH/GSSG, the Superoxide Dismutases, the Glutathione peroxidases, the Catalase). The effect of OS on biomolecules appears on proteins, DNA and lipids as shown in the hexagon in the middle of the figure, with specific chemical modifications. Below, the placenta on which this review is focused is presented, part of the oxidative stress response being triggered in this organ by the activation of the P38/MAPK pathway. In black are the sources of oxidative stress in the human placenta that are described in this paper (Metalloids and Metals, Tobacco smoke and Atmospheric particulate matter). In Blue boxes at the right part of the figure are presented specific chemicals that are part of the placental regulation of oxidative stress, acting either on ROS production or modulation of ROS detoxification that is further detailed in the text. Other abbreviations: ETC = Electron Transport Chain, NOX = Nitric Oxidases, sFLT1 = Soluble FMS-Like Tyrosine kinase 1, sENG = soluble Endoglin, CO = Carbon Monoxide, TOCP = Tri-ortho-cresyl phosphate.
3. Final Considerations and Conclusions
Herein attempted to connect three concepts: (1) environmental exposure to contaminants, (2) the induction of oxidative stress in the placenta and (3) obstetric diseases of placental origin. The mechanistic studies that we present seem to demonstrate that the impact of environmental pollutants upon the health risk is genuine and that the generation of oxidative stress in the placenta is a sensible and truthful mediator of placental diseases. As such, in the STOX1 mouse model of preeclampsia, we have shown previously that oxidative/nitrosative stress is a major cause of the onset of the disease [57][58][153,154], corroborating the importance of this pathway in hypertensive disorders of pregnancy [59][60][155,156]. Specific environmental pollutants are now produced en masse for novel technological developments, for instance, connected to the upsurge of electric vehicles in many countries, leading to an exponential production of batteries that may release novel molecules in our environment. These batteries depend upon the use of rare elements (neodymium, lanthanum, terbium, dysprosium, lithium, cobalt that may reveal their effect on placental health in the future). For example, an ancient German publication revealed that in highly contaminated environments, traces of lanthanum and bromide are detectable in the placentas [61][157]. There is more recent literature regarding lithium exposure, showing, for instance, in an Argentinian mother-child cohort, that elevated exposure to this metal as well as to boron, arsenic, and antimony induced a reduction of the telomere length in the placenta (especially through arsenic exposure, while lithium apparently induced elongation of the telomeres in maternal tissues [62][158]). Earlier, lithium in the maternal blood was shown negatively associated with all fetal measures of size [63][159]. By itself, lithium has been recurrently associated with oxidative stress, including in placental cells [64][160]. Cobalt is also a potential risk factor since exposure to this metal in cells mimics hypoxia, by itself a potential cause of oxidative stress. Besides these technological novelties in transportation, other industrial developments linked to ecological concerns, such as recycling activities (for instance, of electronic material, called ‘e-waste’), increase the risk of propagating novel molecules in the atmosphere. Recently, a meta-analysis revealed that amongst 20 studies [65][161], one explored DNA damage in the placenta [66][162] and found reduced telomere length in this organ; in this study, cadmium placental concentrations were associated with the phenotype; since oxidative stress (such as caused by cadmium exposure) is linked to telomer attrition [67][163], this is a possible mechanism, albeit not directly studied in the paper. The authors surmised that improper e-waste processing was the primary cause of the exposure. In the future, such exposition may gain in importance. Other important sources that can be expected are flame retardants such as polybrominated biphenyl esters (PBDE, [68][69][164,165]), polychlorobiphenyls (PCBs), or other molecules. Overall, recycling consequences of e-waste exposure in China have been reviewed, including in the placenta [70][166]. Plausible health consequences remain an important preoccupation for the future. In conclusion, the induction of oxidative stress in the placenta by environmental toxicants has been studied in a very limited number of cases (as summarized in Figure 23). Similar to other tissues or organs, these studies point to the mitochondria and the cellular antioxidant activity (both enzymatic and non-enzymatic) as main targets. However, to design effective therapeutic approaches, more studies using in vivo and in vitro models are required to investigate the precise mechanisms involved in the induction of oxidative stress by at least the principal pollutants known to impact pregnancy.
Figure 23. A brief graphic summary of the mechanistic impacts of environmental exposure through oxidative stress alterations in the human placenta, as detailed in the text of this review. At the center of this graph is the mitochondria which is the major producer of oxidative stress (ROS = Reactive Oxygen Species, and RNS = Reactive Nitrogen Species). The red dashed arrows relate to the generation of oxidative stress; the box on the upper left summarizes the antioxidant mechanisms that will fight against oxidative stress (the balance GSH/GSSG, the Superoxide Dismutases, the Glutathione peroxidases, the Catalase). The effect of OS on biomolecules appears on proteins, DNA and lipids as shown in the hexagon in the middle of the figure, with specific chemical modifications. Below, the placenta on which this review is focused is presented, part of the oxidative stress response being triggered in this organ by the activation of the P38/MAPK pathway. In black are the sources of oxidative stress in the human placenta that are described in this paper (Metalloids and Metals, Tobacco smoke and Atmospheric particulate matter). In Blue boxes at the right part of the figure are presented specific chemicals that are part of the placental regulation of oxidative stress, acting either on ROS production or modulation of ROS detoxification that is further detailed in the text. Other abbreviations: ETC = Electron Transport Chain, NOX = Nitric Oxidases, sFLT1 = Soluble FMS-Like Tyrosine kinase 1, sENG = soluble Endoglin, CO = Carbon Monoxide, TOCP = Tri-ortho-cresyl phosphate.