1. Introduction
The discovery of antibiotics has drastically changed modern medicine and extended the human lifespan. The first naturally occurring antibiotic, namely penicillin, was discovered by Alexander Fleming from
Penicillium notatum in 1928, which was introduced into clinical practice in 1941
[1][2][8,9]. Most of the antibiotics that are used today were discovered and introduced into the market before 1987, and this period is often termed as the golden decade
[3][10]. Particularly, the cessation of new antimicrobial classes was apparent from 1987 until today
[4][11]; during this period, only a few (~9) new antimicrobial classes were discovered and launched into the markets (
Figure 1). With this slow rate of new antibiotic discovery and the rapid emergence of resistant bacteria, we are in the post-antibiotic era
[5][12].
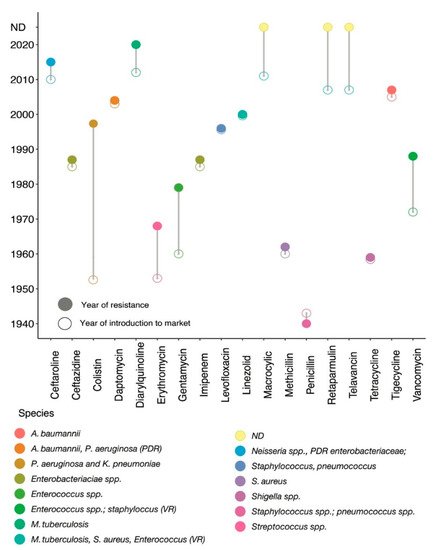
Figure 1. Historical panorama of antibiotic launch and resistance detection. The x-axis indicates different types of antibiotics and the corresponding y-axis shows the year of introduction into clinical practices. Resistance histories to different antibiotics are shown by different circles. Connecting line between empty and filled coloured circles shows the year of introduction of a specific antibiotic into clinical practice and the year of resistance observed for that antibiotic; each coloured circle further represents different bacterial species. For example, colistin was first introduced into clinical practice in 1952
[6][13], but resistance to colistin was first reported in clinical
P. aeruginosa and
K. pneumoniae (shown by a specific coloured circle) in 1998
[7][14]. Penicillin resistant laboratory
E. coli was reported in 1950
[8][15] before its introduction into clinical practice in 1941, but the first penicillin resistance clinical
S. aureus was reported in 1942
[9][10][16,17]. PDR: pan-drug resistant; VR: vancomycin resistant; spp: species; ND: resistance mechanism not detected.
2. Antibiotics and Their Impact on Bacterial Cellular Perturbation
Antibiotics belong to different classes of chemicals—including those of biological, synthetic, or semi-synthetic origin—and have selective modes of action. Based on their mechanisms of action, antimicrobial compounds are classified into two groups: bacteriostatic (
Table 1) and bactericidal (
Table 2). Bacteriostatic drugs are only able to inhibit or hinder growth but cannot kill bacteria. Whereas drugs are called bactericidal when exposure to this group of antibacterial compounds leads to the death of bacteria. Both drugs principally interfere with bacterial cell-wall biosynthesis, DNA synthesis, RNA synthesis, and protein synthesis
[11][18]. Furthermore, antibiotics have been classified based on the cellular components or systems they affect in bacteria. Some of these antimicrobial agents target the synthesis of important cellular components, whereas some other classes of antibiotics interfere with bacterial nucleic acid synthesis or repair
[12][13][19,20]. The mechanistic actions of the bacteriostatic and bactericidal are featured in
Table 1 and
Table 2. Furthermore, we have described the mechanistic action of the major bactericidal antibiotics in the following sections.
Table 1. Action and common resistance mechanisms of major bacteriostatic antibiotics.
|
| Bacteriostatic Candidates |
|
| Mode of Action |
|
Action and common resistance mechanisms of major bactericidal antibiotics.
|
| Bactericide Candidates |
| Mechanism of Resistance |
|
|
| Mode of Action |
|
| Mechanism of Resistance |
|
|
| Tetracycline |
|
| Reversibly inhibits 30S ribosomal subunit of bacteria [14][25]. |
|
| Efflux system and protecting ribosomes [15][26]. |
|
, altered PBP2a encoded by | mecA | [7][29][14,40], extended-spectrum-beta-lactamases (ESBLs), AmpC beta-lactamase (i.e., blaAmpC) |
| Macrolides |
|
| Reversibly inhibits 50S ribosomal subunit of bacteria [16][27]. |
|
| Methylation of the 23S rRNA, efflux system [17][28]. |
|
[ | 30 |
36 |
] |
. |
|
|
| Trimethoprim |
|
| Occupying the active site of bacterial dihydrofolate reductase (DHFR), thus blocking the activity of the enzyme [26][37]. |
|
|
| Penicillins |
|
| Competitively inhibits the transpeptidase enzyme resulting cross-linking blockage in cell wall [28][39]. |
|
| Beta-lactamase encoded by blaZ][31][32][41,42,43]. |
|
|
| Cephalosporins |
|
| Competitively inhibits the transpeptidase enzyme resulting in cross-linking or blockage in cell wall [33][44]. |
|
| AmpC beta-lactamase (i.e., blaAmpC), ESBLs (i.e., blaCTX-M) [30][31][41,42]. |
|
|
| Sulphonamides |
|
| Inhibits folate synthesizing enzyme dihydropteroate synthase (DHPS) [18][29]. |
|
| By horizontal transfer of dihydropteroate synthase gene [19][ |
|
| Carbapenems |
|
| Binding with penicillin-binding proteins (PBPs) and inactivation of these proteins leads to cell wall synthesis interruption [34][45]30]. |
|
. |
|
| Carbapenemases (i.e., class A serine-carbapenemase including KPCs; class B metallo-carbapenemase including New-Delhi-metallo-beta-lactamases or NDM, Verona-integron-encoded beta-lactamases or VIM, Imepenemase IMP-carbapenemase (also a metallo-beta-lactamase); class D serine carbapenemase such oxacillinase (OXA) [35][36][46,47], mutation-derived target enzyme modification [37][48]; preventing the drug entry by modifying outer membrane permeability [38][49]; pumping carbapenems out by efflux pump systems [39][50]. |
|
| Streptogramins |
|
| Reversibly inhibits 50S ribosomal subunit of bacteria [20][31]. |
|
| Acetyltransferases vatD gene expression mediates streptogramin A, wheras vatE and ermB or vgbA gene cluster confers streptogramin B antibiotics [21][32]. |
|
|
| Oxazolidinones |
|
| Reversibly inhibits 50S ribosomal subunit of bacteria [22][33]. |
|
| High diversity and coselection of optrA [23][ |
|
| Aminoglycoside |
|
| Binding with 30 s ribosomal subunit resulting genetic code misreading followed by interruption of bacterial translation [40][51]. |
|
| Mostly through aminoglycosides modifying enzymes encoded by aac (aminoacetyl-tranferase) and aph (aminophospho-transferase), efflux system, or mutation in rpsL and 16S rRNA [32][41][43,52]. |
|
34 |
|
| Fluoroquinolones |
|
| Interrupting bacterial DNA replication by inhibiting topoisomerases [42][53].]. |
|
|
|
| Target enzyme mutation (DNA gyrase encoded by gyrA and gyrB, and topoisomerase IV encoded by parC and parE genes), efflux system and changing drug entry [43][54]. |
|
| Lincosamides |
|
| Reversibly inhibits 50S ribosomal subunit of bacteria [24 |
|
| Rifamycin |
| ][35]. |
|
| Interrupting transcription by inhibiting bacterial RNA polymerase [44][55]. |
| Target site modification, efflux system and drug inactivation [25][ |
| Mutation of the target (beta subunit of RNA polymerase encoded by rpoB) [45][56]. |
|
[ |
|
|
| Polymyxins |
| 46Increase expression of DHFR or decrease the affinity of DHFR to the drug [ |
| Binding to lipid A of LPS and interfere with outer membrane permeability ][27][38]. |
|
Table 2.57 |
] |
. |
|
|
|
|
The |
pmrHFIJKLM | (also known as | arn operon) and pmrCAB operon—both invove in the biosynthesis of LAra4N and modify the lipid A, thus disrupt lipid A charges [10][17]; mutations in genes encoding the two-component regulatory systems such as pmrAB [47][58], phoPQ and plasmid-borne mcr genes confer resistance to colistin—the last line of drug [48][49][59,60]. |
|
|
| Daptomycin |
|
| Binding to anionic phospholipids in the cytoplasmic membrane [50][61]. |
|
| Mutations in gene mprF which encodes the multiple peptide resistance factor [51][62]. |
|
|
| Vancomycin |
|
| Binding to the dipeptide terminus d-Ala-d-Ala of peptidoglycan pentapeptide precursors preventing peptidoglycan crosslinking leads to the inhibition of bacterial cell wall synthesis [52][63]. |
|
| Replacing d-Ala-d-Ala with d-Ala-d-lac or d-Ala-d-Ser alternatives to which vancomycin has low affinity [53][64]. |
|
A commonly used bactericidal class of antibiotic is fluoroquinolone which targets and inhibits DNA replication by interfering with topoisomerase II enzyme—also known as DNA gyrase composed of two subunits encoded by
gyrA and
gyrB—leading to cellular death by forming double-strand DNA breaks
[54][21]. Whereas beta-lactam antibiotics—including penicillins, cephalosporins, carbapenems, and monobactams—act by binding to and inhibiting the penicillin binding proteins (PBP) leading to stop in cross-linking or transpeptidations within the bacterial cell wall, and thus undergo cellular death
[55][22]. Furthermore, bactericidal antibiotics interfere with common metabolic systems such as the central metabolic pathways called tri-carboxylic acid (TCA) cycle and iron metabolism. For example, reactive oxygen radicals in response to lethal bactericidal antibiotics result in cellular death
[56][57][23,24].
Rifamycin is a semi-synthetic bactericidal class of antibiotic that can induce cell death by inhibiting bacterial RNA synthesis
[58][65]. During the execution of the normal cellular function, beta-subunit of DNA-dependent RNA polymerase enzyme is involved in the stable channel formation between RNA-polymerase and DNA complex from which newly synthesized RNA strand arises
[59][60][66,67]. Rifampicin stably binds to the beta-subunit of DNA dependent RNA polymerase (encoded by
rpoB gene), thus inhibiting the high-fidelity transcription and causing cellular death.
Another class of bactericidal antibiotic such as aminoglycoside causes bacterial cellular death by interfering with cellular energetics, ribosome binding and protein synthesis inhibition
[61][68]. Bacterial protein synthesis through the translation of mRNA occurs in a sequential fashion involving initiation, elongation, and termination. This process is operated in the cytoplasmic space involving the collaborative action of the ribosome (which acts as a factory) and many other important accessory translation factors available in the cytoplasm
[62][69]. The ribosome is composed of two ribonucleoprotein subunits called the 30S (encoded by
rpsL gene) and 50S. Following the formation of a complex between mRNA-transcript, N-formyl methionine-charged aminoacyl tRNA, several initiation factors and a free 30S subunit (this process is called initiation step of translation), the ribosome is assembled for the next translational step
[63][70]. While translation is a complex process that requires many cellular components and translation factors, drugs can interfere with protein synthesis in various ways. Antibiotics that inhibit protein synthesis are classified into the 50S and 30S inhibitors, respectively. The 50S inhibitors (i.e., erythromycin, clindamycin, streptogramin, chloramphenicol, and linezolid) interfere with protein synthesis by blocking the initiation of protein translation or translocation of peptidyl tRNAs
[64][65][71,72]. Inhibition of 30S ribosome (i.e., caused by tetracyclines and aminocyclitols) involves blocking of the access of aminoacyl tRNAs to the ribosome. Both spectinomycin and aminoglycosides—including streptomycin, kanamycin, and gentamycin—bind to the 16S rRNA, a component of the 30S ribosomal subunit. Specifically, aminoglycosides bind to the 16S rRNA, which in turn alter the conformation of the complex formed between an mRNA codon and its cognate charged aminoacyl tRNA in the ribosome. This interaction results in defective protein
[12][15][66][19,26,73], thus cellular death occurs.
Polymyxins group of antibiotics (polymyxin A–E) are of cationic cyclic polypeptide origin with strong bactericidal effects. Among these, only polymyxin B and polymyxin E—also called colistin—are used in clinical practices
[67][74]. Both antibiotics are commonly prescribed to treat infections caused by Gram-negative bacterial pathogens, particularly against extensively drug resistant (XDR)
P. aeruginosa and
A. baumannii [68][75]. These cyclic antimicrobial peptides have long hydrophobic tails which directly interact with LPS of the outer membrane of Gram-negative bacteria. Particularly, polymyxins destabilize calcium and magnesium bridge by binding to the lipid A component of the LPS. This event causes bacterial outer membrane permeabilization and allows polymyxins to enter the outer membrane, leading to cellular death
[49][60].
Daptomycin is another class of bactericidal antibiotic and is composed of cyclic polypeptides. Daptomycin is used for the treatment of infections caused by Gram-positive bacteria, particularly against methicillin resistance
S. aureus and
Enterococcus faecium. Daptomycin interacts with anionic phospholipids in the presence of calcium ions in the cytoplasmic membrane. This interaction helps daptomycin penetrate the membrane, which ultimately causes membrane depolarization and cellular death. However, unlike most other bactericidal antibiotics, the detailed mechanistic basis of cellular death and resistance for daptomycin is not yet fully elucidated
[69][76]. Detailed mechanisms of action of the major classes of bactericidal antibiotics are depicted in
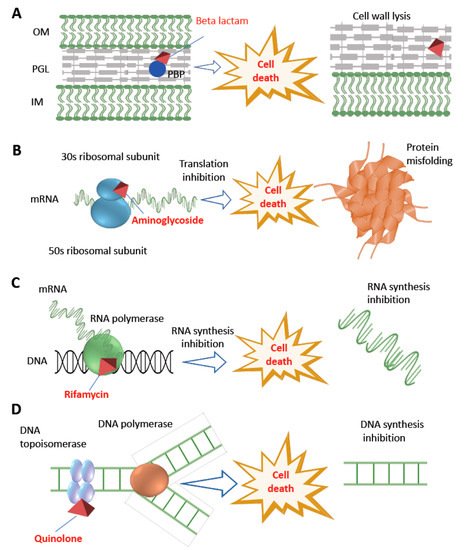
Figure 2.
Figure 2. Diversity of antibiotic resistance mechanisms. The figure shows the major bactericidal antibiotics and their different targets. Beta-lactam antibiotics degrade bacterial cell wall by interfering with cross-linking or transpeptidations within the bacterial cell wall by binding with PBP (panel A), aminoglycoside interferes with protein synthesis by binding with 30S ribosomal subunit (panel B), rifamycin inhibits bacterial transcription by interfering with beta-subunit of DNA dependent RNA polymerase enzyme (panel C), whereas quinolone class of antibiotics inhibit DNA synthesis by interfering with DNA topoisomerase (panel D). OM: outer membrane; PGL: peptidoglycan layer; IM: inner membrane; PBP: penicillin binding protein.