Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Oanh Vu.
G protein-coupled receptors (GPCRs) represent the largest membrane protein family and a significant target class for therapeutics. Receptors from GPCRs’ largest class, class A, influence virtually every aspect of human physiology. About 45% of the members of this family endogenously bind flexible peptides or peptides segments within larger protein ligands. While many of these peptides have been structurally characterized in their solution state, the few studies of peptides in their receptor-bound state suggest that these peptides interact with a shared set of residues and undergo significant conformational changes.
- peptide GPCR
- class A GPCR
- peptide docking
- non-canonical amino acids
1. Introduction
1.1. G Protein-Coupled Receptors Are a Significant Target of Therapeutic Intervention
With more than 800 members, G protein-coupled receptors (GPCRs) are the largest family of human transmembrane proteins [1]. They are key players in many physiological functions, regulate the majority of cellular processes, and are involved in numerous disease pathologies [2]. By subtracting the olfactory/odorant GPCRs involved in recognizing smells, about 400 human GPCRs are considered as druggable. Their substantial involvement in cellular signaling has established GPCRs as highly relevant pharmacological drug targets. About 34% of all drugs approved by the US Food and Drug Administration (FDA) achieve their therapeutic effects through GPCRs [3].
1.2. Peptide-Activated Receptors Are a Large Percentage of the GPCR Class A
Out of four classes of GPCR—A, B, C or F—Class A is the largest and most diverse group in humans. This subfamily has been investigated most extensively in drug discovery due to their available structural and experimental data. They conform with the common GPCR structural fold, such as a seven-transmembrane (7TM) helices domain, three extracellular loops, and three intracellular loops with ligand-binding pockets and a G-protein-binding region located in the extracellular and intracellular ends of the helix bundle, respectively [4]. The variety of drugs targeting GPCRs reflect the diversity of chemical signals that can be transduced by GPCRs, including small molecules, lipids, ions, and proteins [5,6][5][6]. In particular, according to the data from the GPCRdb server [4], the peptide- and protein-activated receptors are found to account for about 46% of all class A GPCRs in humans. For this review, we consider GPCRs that recognize classical peptides and peptide-like segments within larger protein domains and belong to the same category of receptors. Peptide-activated receptors are found across all rhodopsin-like subfamilies (α, β, γ, and δ) and the entire secretin family [7]. Given this coverage, it is unsurprising that many of the aforementioned blockbuster drugs (e.g., olmesartan, buserelin, and valsartan) target members of this receptor group. While Olmesartan and Valsartan serve as an angiotensin II receptor blocker (ARB) in treating hypertension [8,9][8][9], buserelin, a luteinizing hormone—releasing the hormone (LHRH) agonist, can be used to treat hormone responsive cancers, such as prostate and breast cancer [10]. In 2020, nearly 50 GPCR peptide drugs have been approved [11]. In accordance with this importance for therapeutic development, a full understanding of the structural and dynamical determinants of signaling for these molecules is necessary. This review covers what is known regarding these receptors structurally using various biophysical techniques and provides suggestions for future discovery routes.
1.3. Diversity of Peptide Ligands
Peptide ligands come in a variety of lengths and structures, although they share the common theme that they are ribosomally translated. Often, these peptide ligands are produced as pre-hormones that are subsequently processed to their active form. As a result, peptide ligands range in size from three amino acids (e.g., thyrotropin-releasing hormone (TRH)) up to ~100 amino acids (e.g., chemokine ligand 23 (CCL23)). In addition to size differences, many peptide hormones undergo post-translational modifications. Some of these modifications are necessary to increase the peptide half-life by inhibiting exopeptidases, such as N-terminal pyroglutamation (e.g., TRH and luteinizing hormone (LH) [12]) and C-terminal amidation (e.g., neuropeptide Y (NPY), pancreatic polypeptide (PP), and peptide YY (PYY) [13]). However, in some cases, these modifications serve dual purposes by acting as molecular recognition sites in their cognate receptors [14]. Other types of post-translational modifications include lipidation, bromination, and disulfide bridge formation. A summary of modifications is found in Table 1. These modifications further increase the diversity of chemical space available to peptide hormones beyond the canonical 20 amino acids. The size, sequence, shape, charge, structural dynamics, and chemical diversity allow for a vast degree of specificity between peptide hormones and their receptors. Furthermore, it is common for a given peptide hormone to exist in multiple isoforms, such as the neuropeptide Y (NPY) family, which consists of NPY, peptide YY (PYY), and pancreatic polypeptide (PP) and the endothelin peptides ET-1, ET-2, and ET-3.
Table 1. Examples of peptide hormone modifications.
| Peptide Modification | Example | Function |
|---|---|---|
| C-terminal amidation | Neuropeptide Y (NPY), neuromedin B | C-terminal amidation reduces the overall charge of a peptide, forms key hydrogen interactions that are important for the potency of the peptide [15], and increases the metabolic stability of peptides as well as their ability to resist enzymatic degradation [16] |
| N-terminal pyroglutamic acid | Thyroid stimulating hormone (TSH), gonadotropin-releasing hormone I (GnRHI), regulated upon activation, Normal T cell expressed, and presumably secreted (RANTES)/chemokine ligand 5 (CCL5) | The pyroglutamic acid is often involved in peptide-receptor recognition and potency [17], and provides stability against N-terminal degradation [18] |
| Bromination | Neuropeptides B and W (NPBW) | Bromination on N-terminal tryptophan might protect the peptide from amino-peptidases’ degradation [19] |
| Lipidation | Ghrelin | The attached lipid group (e.g., octanoyl group) is essential to the activity of the peptide [19] and affects the hydrophobicity of the peptide [20] |
| Disulfide bridge formation | Endothelin, vasopressin | The disulfide bonds stabilize the defined secondary structure [21], stabilizing the bound conformation of the peptide [22] |
| Differential proteolysis | Bradykinin, angiotensin, NPY/NPY3-36, apelin (Ape)-13/Ape-17/Ape 36, adrenocorticotropic hormone (ACTH), pro-opiomelanocortin (POMC) cleavage yielding α-, β-, and γ- melanocyte-stimulating hormone (MSH), and endorphins | Proteolysis can switch the activity of the peptides on and off [23] or differentiates the binding selectivity and the biological responses of the peptides [24] |
1.4. Reducing the Flexibility of Peptide Ligands Is Crucial for Success in Co-Crystallization
A significant challenge for the interpretation of structures determined via crystallization of peptide-activated receptors in complex with their cognate peptide ligand is the peptides’ inherent flexibility. Typically, small molecule antagonists and agonists will adopt a single conformation when interacting with a receptor and are fully encased in the receptor-binding pocket. Peptide ligands may adopt a single conformation in the binding pocket. However, due to their length, the remainder of the ligand can remain outside the binding pocket and be flexible. This conformation change is likely the reason that neurotensin 1 receptor (NTS1R) was crystallized with only residues 8–13 of the peptide, since residues 1–7 are expected to extend above the receptor pocket and remain unconstrained [25,26][25][26]. The peptide ligand of the apelin receptor, while full-length, was modified to incorporate a lactam ring, which significantly constrained the peptide’s flexibility [27]. Full-length chemokine crystallization is possible, as the portion of the chemokine that extends out of the binding pocket folds into a well-defined structural domain. However, the N-terminus of the receptor, known to recruit and bind the chemokines, has yet to be determined experimentally in its entirety [28,29,30][28][29][30].
1.5. Complexity of Peptide Ligand and Receptor Interactions
In addition, as was recently classified, many peptide ligands target multiple receptors adding to their signaling complexity [2]. This complex selectivity of peptide ligand/receptor interactions results in the peptide ligand biology’s common theme: Multi-ligand/multi-receptor systems. To date, evidence shows that the related ligands binding to the same receptor or the same ligand binding to two different receptors can adopt different bound state conformations and sustain deviating interaction networks [31[31][32],32], activating the receptors by the induced-fit or conformational selection. However, the system of multi-ligand/multi-receptor binding is different from the promiscuous binding of major histocompatibility complex (MHC) molecules to antigenic peptides. The GPCR-peptide binding relies on the conservation of residue pairwise interactions among evolutionarily related peptides and GPCRs receptors. In contrast, MHCs, due to their conformational flexibility, can fold into multiple active states to bind to a diverse set of antigenic peptides. A precedent kinetic study observed a slow rate, suggesting that the mutual configurational complementarity took time to be sufficient for flexible MHC and flexible peptides in order to form an initial complex [33]. This theme of multi-ligand/multi-receptor systems complicates the formulation of overarching binding and activation mechanisms that holistically explain this category of receptors, unlike what is known regarding receptors activated by bioamines [34,35,36][34][35][36]. Moreover, it complicates the development of selective probes and therapeutic agents. Therefore, it is critical for a full understanding of receptor/hormone biology to study each peptide ligand/receptor combination in detail before attempting to formulate generalizations that can be used for future drug development. This task is fundamental through many ongoing efforts in order to achieve this step.
2. Comparison of Peptide Binding Modes across Class A GPCRs
2.1. Diversity in the Binding Modes of the Peptide Ligands to Class A GPCRs
The first crystal structure of a peptide-activated receptor was the CXCR4 receptor in 2010 [37]. The receptor structure was determined in the inactive state bound to both a small molecule antagonist and a peptidomimetic. This receptor structure was similar to what had previously been seen for aminergic [38,39][38][39] and nucleotide [40] receptors. However, an interesting difference was the presence of an β-hairpin in extracellular loop 2 (ECL2), a motif that has been present in all peptide-activated receptor structures reported since that time [41].
Moreover, two additional years passed before another peptide-activated receptor structure was determined. The year 2012 was a watershed year for this family with the structure determination of all four opioid receptor (OR) members (δOR [42], κOR [43], µOR [44], and NOP [45]), the protease-activated receptor type 1 (PAR1) [46], and the neurotensin type 1 receptor (NTS1R) [15]. Notably, the NTS1R structure was the first structure that was determined as a peptide-activated receptor in complex with its endogenous peptide ligand. Interestingly, NT’s binding depth was not as pronounced as seen for the aminergic and nucleotide ligands, suggesting that peptide ligands bind more superficially and predominantly interact with the extracellular loops. As the extracellular loops are the most divergent region of GPCRs, this prevented the extrapolation of this binding mode to other peptide ligands.
Since 2012, additional peptide-activated receptor structures were determined. These included further chemokine receptors (CCR2 [47], CCR5 [48], CCR9 [49], and the viral US28 chemokine receptor [18]), both subtypes of the orexin [50,51][50][51] and angiotensin [52,53][52][53] receptors, the PAR2 receptor [54], the endothelin-B receptor [55], the neuropeptide Y type 1 receptor [56], the neurokinin 1 receptor [57], and the C5a receptor [58]. The binding pockets of peptide-activated GPCRs are uniformly wide due to the structured ECL2 but display a variety of hydrophobic and electrostatic conditions [42]. Of note, only a small subset of these structures has been determined with a peptide ligand bound. These include the chemokine receptors US28, CCR5, and CXCR4 [18,19,59,60][18][19][59][60], the endothelin-B receptor [55], the apelin receptor [17], the µ opioid receptor [61], the angiotensin type II receptor [53], and the C5a receptor [62].
In contrast to the observed orientation of NT(8-13), these ligands’ binding modes are very diverse, as seen in Figure 1. Peptide ligands can unwind their helix and adopt unstructured conformations to penetrate deep in the helical bundle via their N- or C-terminus, such as apelin. This observation of great diversity in peptide binding modes among GPCRs was also confirmed by the previous review work [63]. They can bind with both termini folded into the binding pocket, such as ET-1 or in a horseshoe manner, presenting a curved surface to the receptor, such as gp120. The ligands can bind deeply (sAngII, DAMGO, ET-1, and vMIP-II) or closer to the surface (gp120, CX3CL1, PMX53, and 5P7-CCL5). However, conservation in the peptide engagement mechanism among class A GPCRs has been investigated by combining earlier SAR studies and the alignment of interacting residues from recent GPCR-peptide structures. The authors suggested that common patterns in peptide-GPCR interactions were divided into four groups, depending on whether the peptide is cyclic or not and whether the GPCR interacts with the N- or the C-terminus of the peptide [64]. By superimposing the structures of the complexes, a common observation between the binding modes of different peptide ligands is that they often bind over an extended surface of the receptor (Figure 2A). More interestingly, we notice that peptides align surprisingly well at the core of the binding pocket (Figure 2B). Together with the conserved β-hairpin in ECL2, these observations suggest potential general themes conserved within GPCRs binding peptide-ligands.
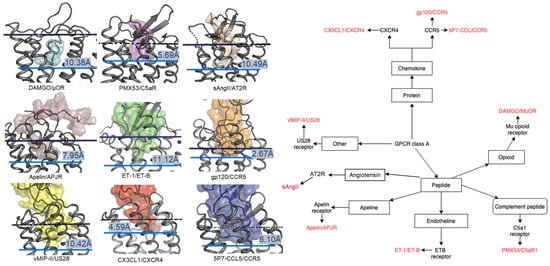
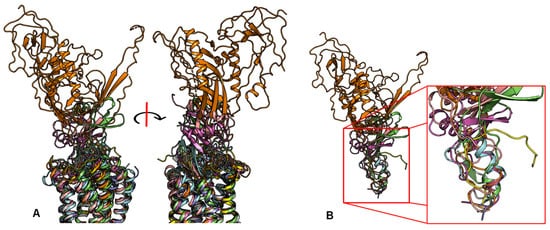
Figure 1. Overview of nine co-crystal structures of class A peptide-GPCR. (Left) Comparison of peptide binding modes and crystallized peptides DAMGO (cyan), PMX53 (magenta), sAngII (beige), apelin derivative (salmon), ET-1 (green), gp120 (orange), vMIP-II (yellow), CX3CL1 (red), and 5P7-CCL5 (blue) at the receptors µ opioid receptor (µOR) (PDB ID: 6DDE), complement component 5a receptor (C5aR) (PDB ID: 6C1R), angiotensin II type 2 receptor (AT2R) (PDB ID: 5XJM), apelin receptor (APJR) (PDB ID: 5VBL), endothelin B receptor (ET-B) (PDB ID: 5GLH), C-C chemokine receptor type 5 (CCR5) (PDB ID: 6MEO), US28 (PDB ID: 4XT1), CXC-chemokine receptor 4 (CXCR4) (PDB ID: 4RWS), and CCR5 (PDB ID: 5UIW), respectively [22,32,34,37,38,39,40,41,42][22][32][34][37][38][39][40][41][42]. All receptors were aligned in the transmembrane region. The approximated extracellular border of the transmembrane region is marked in the upper dotted dark blue lines. The membrane region of GPCR receptors was calculated using the PPM server [65]. The lower blue bars and texts illustrate the depth of penetration for each peptide ligand. (Right) Classification tree of eight class A GPCRs with their nine peptide ligands in those nine listed structures.
Figure 2. Despite the diversity in the peptide engagement, their overlapping region at the core of their binding pocket suggests common ligand-GPCR interactions. (A) Superimposition of the nine peptides/class A GPCR complexes. (B) Overlay of all peptide ligands and zoom-in of the peptide region at the cores of GPCRs.
2.2. Peptide Ligands Affect the Conformation of the Extracellular Surface
An essential consequence of the extended binding surface area of peptide ligands is that their presence affects not only the deep binding pocket, but also the extracellular loops. This link between ligand engagement and GPCR loop conformation was recently demonstrated by the endothelin receptor structures [55]. This receptor was crystallized in the apo state and in complex with a peptide ligand. Interestingly, there was an extensive rearrangement of the extracellular domain in the peptide ligand presence (Figure 3A). This conformation rearrangement is expected to be the case for many peptide-activated receptor structures. In particular, the structural model of the Y1 receptor in complex with a small ligand found the N-terminus of the receptor lying over the binding pocket [56]. Mutagenesis studies confirmed that this portion of the receptor did not affect the binding properties of the small molecule or endogenous peptide. It was implied that the N-terminus needed to be displaced from this crystallized orientation to allow for the binding of the considerably larger NPY ligand (Figure 3B). This implication was modeled and presented with the crystal structure with an extensive use of orthogonal biophysical techniques, including NMR, cross-linking mass spectrometry, and mutagenesis. Additionally, the structure of the AT1R with a small molecule antagonist found the N-terminus lying over the ligand-binding pocket. In contrast, the AT2R structure, which was determined in the presence of a peptide analog sAngII, required the N-terminus to shift to allow for the access of the ligand to the orthosteric pocket (Figure 3C).
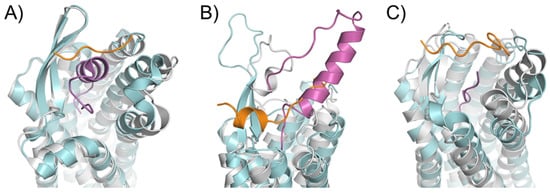
Figure 3. Rearrangements in the extracellular domain of peptide-activated GPCRs for peptide binding. (A) In the apo ET-B receptor (grey, PDB ID 5GLI), the N-terminus (orange) is lying over the ligand binding pocket. In the ET-1-bound state (cyan, PDB ID 5GLH), the bound ET-1 ligand (magenta) occupies the space of the N-terminus leading to its displacement [55]. (B) The crystal structure of antagonist-bound Y1 receptor (grey, PDB ID 5ZBQ) is also found with the N-terminus (orange) lying over the ligand binding pocket. The modeled peptide-bound Y1R (cyan) places the NPY ligand (magenta) in this space displacing the N-terminus [56]. (C) In the antagonist bound AT1 receptor (grey), the N-terminus (orange) extends over the pocket towards ECL2 [66]. In the AT2 receptor (cyan) bound to sAngII (magenta), the peptide binds deep within the pocket and the N-terminus lies over ECL3 [53].
2.3. ECL1 and ECL2 Bound Conformation Have Conversed across Class A Peptide-GPCRs
The superimposition of the three extracellular loops of peptide GPCR class A shows that the bound conformations of ECL1 and ECL2 are considerably more conserved than the ECL3 (Figure 4). This observation sugsgests that the first two extracellular loops could support a general interface for peptide binding. Together with the conserved β-hairpin in ECL2, details of the ECL local structure and orientation are critical for recognizing specific peptide ligands.
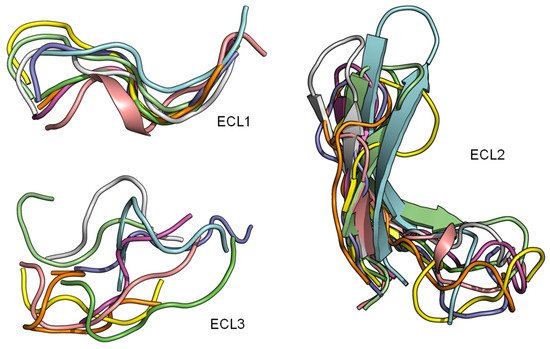
Figure 4. ECL1 and ECL2 have a conserved bound conformation compared to ECL3. Overlay of three extracellular loops.
Among the nine class A GPCR structures that we investigated, four ECL1s have a common motif Y/HxWxF, and eight of them possess an xWxF motif. This motif, together with residue 2.60, interacts favorably with the bound conformation of the peptides. More specifically, the aromatic Y/H sidechain tends to form hydrogen bonds or hydrophobic interactions with the adjacent peptide sidechain or backbones. Moreover, the residue F23.52 forms an π-π interaction to stabilize the conformation of W23.50, while W23.50 interacts with the peptide directly through hydrophobic interactions or indirectly through an π-π interaction with the nearby W/L2.60 residue (Figure 5-Left). Moreover, we quantified the strength of the aforementioned interactions by computing the per-residue ΔΔG with Rosetta on the contacting GPCR residues. A ΔΔG score is defined as the sum of the “two body” interaction scoring terms between each GPCR and peptide residues. We used the Rosetta Energy Function 2015 or REF2015, which encompasses a mix of weighted physics-based and knowledge-based scoring terms that were designed to evaluate the biomolecular structure, stability, and association. A previous publication has described the mathematical models and physical concepts that underlie the latest Rosetta energy function [67]. Supplementary Table S1 lists the two-body scoring terms that were included in the calculation of ΔΔG scores. The ΔΔG values of the three key residues (Y/H, W, and F) of the Y/HxWxF motif, together with the residue 2.60, are indicated by the colors on the images and reported in the table in Figure 5. In general, the ΔΔG values for those residues are negative, suggesting a favorable interaction energy. This quantitative analysis further confirms our observations regarding the common ECL1 mode of peptide engagement among the nine class A GPCR structures.
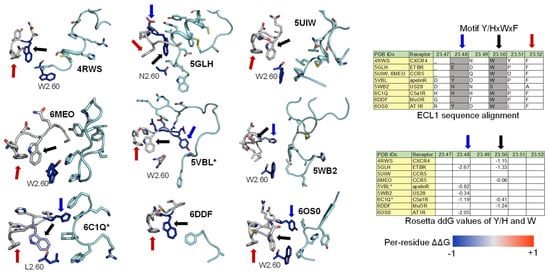
Figure 5. ECL1 and the role of motif Y/HxWxF in peptide binding among class A GPCRs with peptide ligands. (Left) Interactions among ECL1, residue 2.60, and the peptide. The interacting peptide residues are colored in cyan. Residues on ECL1 and 2.60 are colored based on their computed per-residue ΔΔG values (blue: Negative ΔΔG, darkest blue: −1 or below; grey: ΔΔG value of 0 or no interactions; red: Positive ΔΔG, darkest red: 1 and above). (Right) The tables show the sequence alignment of ECL1 and the three key residues in ECL1 motif Y/HxWxF are Y/H, W23.50, and F23.52, which are marked with blue, black, and red arrows, respectively.
Similarly, we conducted the ΔΔG analysis on ECL2 residues and observed that all peptides interact favorably with the β-hairpin of this loop. Out of the three extracellular loops, ECL2 tends to be the most structured with a distinctive secondary structure of a twisted beta-hairpin conformation. In all structures of the nine complexes, ECL2 loops maintain the “open” conformation [68], opening a “gate” and allowing the peptide ligand to enter the core of the TM bundle from the extracellular region. Naturally, the peptides would interact with the β-hairpin of ECL2 β-hairpins at the “gate”, which connects the extracellular space to the inside transmembrane domain. This observation is also reflected in the computed ΔΔG of the interacting target residues. The ΔΔG analysis results suggest that the peptides generally engage with the β-hairpin of ECL2, especially at the tip where the three conserved residues (45.50, 45.51, and 45.52) are located (Figure 6 and Figure 7).
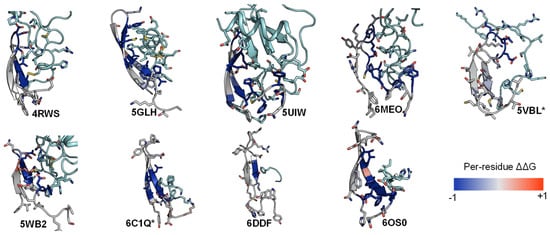
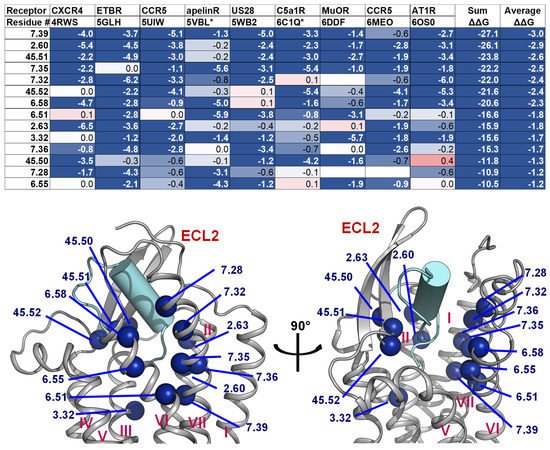
Figure 6. ECL2 β-hairpin and conserved residues interact with peptides of nine peptide/class A GPCR crystal structures.
Figure 7. Residues with the strongest interactions according to the average computed ΔΔG suggest the common binding pocket of peptide ligands. (Upper) A table shows a list of residues with the top average computed ΔΔG values. The residues are numbered according on the Ballesteros-Weinstein numbering scheme [69]. For each residue position, the ΔΔG values are colored in the scale from −1 and less (blue) to 0 (white) to 1 and above (red). The absence of the ΔΔG values indicates that the corresponding residues do not interact with the peptide ligands. Two final columns of the table contain the sum and the average ΔΔG values across nine peptide-class A GPCR structures, respectively. The residue list is sorted in their ascending average ΔΔG order. (Lower) Front and side view of the common peptide binding pocket towards the core of nine class A GPCR structures. The top residues in the upper table are mapped on the ET-1/ETB structure (PDB ID: 5GLH) [55]. The important residues for peptide engagement across eight class A GPCRs are marked by blue spheres. The peptide ligand ET-1 is shown as a cyan cylinder with two unstructured extended regions.
2.4. A List of 14 Common Interacting Residues Suggests a General Peptide Recognition and Binding Mechanism among Nine Class A GPCRs
Despite a considerable diversity in size, sequence, secondary, and tertiary structure of the nine peptide ligands, we observed a significant overlap in the receptor region they bind to, particularly in a binding pocket between the outer leaflet portions of transmembrane helices (Figure 2). Using Rosetta [70[70][71],71], we calculated the per-residue ΔΔG of the interacting residues on the transmembrane helices, two conserved ECL1 residues (23.49 and 23.50), and three conserved ECL2 residues (45.50, 45.51, and 45.52). The details of the structure optimization and ΔΔG analysis protocols are listed in the Supplementary Material, and the ΔΔG values of all residues are listed in the Supplement Table S1. To make the optimization and ΔΔG analysis possible for the apelin/ApelinR (PDB ID: 5VBL) and the PMX53/C5aR (PDB ID: 6C1Q) structures, which contain a non-nature peptide backbone, we generated 5VBL* and 6C1Q* as natural-backbone peptide analogs of those structures. More specifically, the 5VBL* peptide ligand has the native apelin sequence, and the covalent bond between ornithine (ORN) at position 2 and the N-terminal acetyl group is omitted in the 6C1Q* peptide (Figure S3). Then, the GPCR residues were ranked based on their calculated ΔΔG. We selected 14 common residues with ΔΔG of less than −1 and contact peptide ligands in at least seven out of nine GPCR-peptide complexes. The details of the list and their locations on a GPCR structure are mapped in the structure of the ET-1/ETB receptor complex, as shown in Figure 6. This list of the top 14 residues implies a potential common peptide-binding mechanism among class A GPCRs. This common binding pocket encompasses two residues of TM2 (2.60 and 2.63), one from TM3 (3.32), three from TM6 (6.51, 6.55, and 6.58), five from TM7 (7.28, 7.32, 7.35, 7.36, and 7.39), and all three conserved ECL2 residues. More specifically, the common peptide engagement mechanism starts from the end of the β-hairpin of ECL2, extends to the tip of TM2, touches the extracellular half of TM7 and TM6, then ends at the core of TM3. The Supplementary Table S2 summarizes the non-Van Der Waal interactions between these 14 residues and the corresponding peptides. Although additional structures of peptide-GPCR complexes are still needed to validate our hypothesis of the common peptide binding pocket, this finding could help guide future structural studies of this family of GPCRs.
Herein, we examine whether the common binding mechanism agrees with the models of three class A GPCRs—Y1 [56], Y2 [14], and Ghrelin receptor [72]—and their endogenous peptide ligands—NPY and Ghrelin. In those studies, the peptide docking experiments were conducted using FlexPepDock [73] with constraints from mutagenesis, cross-linking, and NMR data. For each complex, the ΔΔG analysis was performed on an ensemble of docking models. The per-residue ΔΔG values were assigned to the interacting residues of the GPCR targets. The peptides’ binding pockets contain all of the 14 common residues, except for ghrelin, which does not contact the residue 7.36. Furthermore, most of the interactions between the common residues and NPY or ghrelin are favorable or at least neutral, except for the high ΔΔG value of residue 7.32 from Y2 (Figure 8). These results imply that the observation of the common peptide engagement pocket can also be applied to the docking study of peptide class A GPCRs, especially with limited experimental data.
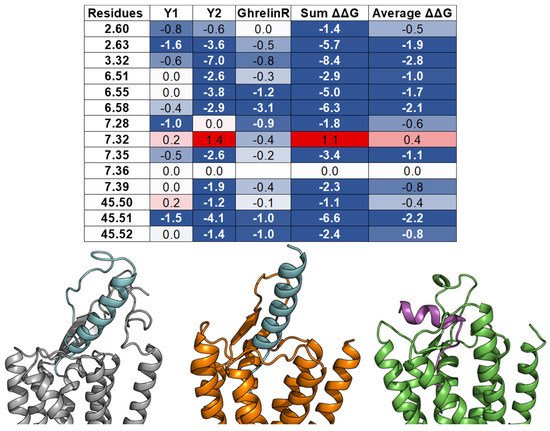
Figure 8. Models of peptide/class A GPCR complexes show that the peptides interact with the top 14 common residues. (From left to right): A table lists the ΔΔGs values of the 14 common residues of Y1 [56], Y2 [14], and ghrelin receptors [72], as well as their sum and average values. The absence of the ΔΔG values indicates that the corresponding residues do not interact with the peptide ligands. The residue ΔΔG cells are colored based on the ΔΔG values (negative: Blue, neutral: White, and positive: Red). The blank cells indicate that the residues do not interact with the peptide ligands. Models of NPY (cyan) bind with the Y1 receptor (grey) and the Y2 receptor (orange), and ghrelin (magenta) binds with the ghrelin receptor (green).
A GPCR pharmacogenomics study has extracted polymorphism data for the coding-region of the 108 GPCR drug targets [74]. From the data provided by the authors, we found around 30 relevant GPCR mutants that were predicted to be deleterious by the sorting intolerant from tolerant (SIFT) [75] or Polyphen [76]. Those 30 genetic invariants have population allele frequencies of around 1 to 28 over 120,000 individuals and are related to the shared peptide interacting residues or are close to those residues. The table containing the information regarding the relevant mutants of peptide and protein binding class A GPCRs is summarized in the Supplementary Table Peptide_binding_pocket_genetic_variants.xlsx. The data suggest the great potential of the proposed common peptide-binding pocket as drug targets for class A GPCRs.
References
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR-G-protein binding. Nature 2017, 545, 317–322.
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842.
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34.
- Isberg, V.; Mordalski, S.; Munk, C.; Rataj, K.; Harpsoe, K.; Hauser, A.S.; Vroling, B.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 2017, 45, 2936.
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427.
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J. 1999, 18, 1723–1729.
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272.
- Scott, L.J.; McCormack, P.L. Olmesartan Medoxomil. Drugs 2008, 68, 1239–1272.
- Markham, A.; Goa, K.L. Valsartan. Drugs 1997, 54, 299–311.
- Brogden, R.N.; Buckley, M.M.T.; Ward, A. Buserelin. Drugs 1990, 39, 399–437.
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413.
- Beck, A.; Bussat, M.C.; Klinguer-Hamour, C.; Goetsch, L.; Aubry, J.P.; Champion, T.; Julien, E.; Haeuw, J.F.; Bonnefoy, J.Y.; Corvaia, N. Stability and CTL activity of N-terminal glutamic acid containing peptides. J. Pept. Res. 2001, 57, 528–538.
- Chance, R.E. Pancreatic Polypeptide. In Encyclopedia of Hormones; Henry, H.L., Norman, A.W., Eds.; Academic Press: New York, NY, USA, 2003; pp. 142–146.
- Kaiser, A.; Muller, P.; Zellmann, T.; Scheidt, H.A.; Thomas, L.; Bosse, M.; Meier, R.; Meiler, J.; Huster, D.; Beck-Sickinger, A.G.; et al. Unwinding of the C-Terminal Residues of Neuropeptide Y is critical for Y2 Receptor Binding and Activation. Angew. Chem. Int. Ed. Engl. 2015, 54, 7446–7449.
- Xu, B.; Vasile, S.; Østergaard, S.; Paulsson, J.F.; Pruner, J.; Åqvist, J.; Wulff, B.S.; Gutiérrez-de-Terán, H.; Larhammar, D. Elucidation of the Binding Mode of the Carboxyterminal Region of Peptide YY to the Human Y2 Receptor. Mol. Pharmacol. 2018, 93, 323–334.
- Bradbury, A.F.; Smyth, D.G. Peptide amidation. Trends Biochem. Sci. 1991, 16, 112–115.
- Goren, H.J.; Bauce, L.G.; Vale, W. Forces and structural limitations of binding of thyrotrophin-releasing factor to the thyrotrophin-releasing receptor: The pyroglutamic acid moiety. Mol. Pharmacol. 1977, 13, 606–614.
- Morty, R.E.; Bulau, P.; Pellé, R.; Wilk, S.; Abe, K. Pyroglutamyl peptidase type I from Trypanosoma brucei: A new virulence factor from African trypanosomes that de-blocks regulatory peptides in the plasma of infected hosts. Biochem. J. 2006, 394, 635–645.
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660.
- Yang, J.; Brown, M.S.; Liang, G.; Grishin, N.V.; Goldstein, J.L. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 2008, 132, 387–396.
- Hewage, C.M.; Jiang, L.; Parkinson, J.A.; Ramage, R.; Sadler, I.H. Solution structure of a novel ETB receptor selective agonist ET1-21 by nuclear magnetic resonance spectroscopy and molecular modelling. J. Pept. Res. 1999, 53, 223–233.
- Hirata, Y.; Yoshimi, H.; Emori, T.; Shichiri, M.; Marumo, F.; Watanabe, T.X.; Kumagaye, S.; Nakajima, K.; Kimura, T.; Sakakibara, S. Receptor binding activity and cytosolic free calcium response by synthetic endothelin analogs in cultured rat vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1989, 160, 228–234.
- Hook, V.; Funkelstein, L.; Lu, D.; Bark, S.; Wegrzyn, J.; Hwang, S.-R. Proteases for processing proneuropeptides into peptide neurotransmitters and hormones. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 393–423.
- Wagner, L.; Wolf, R.; Zeitschel, U.; Rossner, S.; Petersén, Å.; Leavitt, B.R.; Kästner, F.; Rothermundt, M.; Gärtner, U.-T.; Gündel, D.; et al. Proteolytic degradation of neuropeptide Y (NPY) from head to toe: Identification of novel NPY-cleaving peptidases and potential drug interactions in CNS and Periphery. J. Neurochem. 2015, 135, 1019–1037.
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513.
- Da Costa, G.; Bondon, A.; Coutant, J.; Curmi, P.; Monti, J.P. Intermolecular interactions between the neurotensin and the third extracellular loop of human neurotensin 1 receptor. J. Biomol. Struct. Dyn. 2013, 31, 1381–1392.
- Ma, Y.; Yue, Y.; Ma, Y.; Zhang, Q.; Zhou, Q.; Song, Y.; Shen, Y.; Li, X.; Ma, X.; Li, C.; et al. Structural Basis for Apelin Control of the Human Apelin Receptor. Structure 2017, 25, 858–866.
- Burg, J.S.; Ingram, J.R.; Venkatakrishnan, A.J.; Jude, K.M.; Dukkipati, A.; Feinberg, E.N.; Angelini, A.; Waghray, D.; Dror, R.O.; Ploegh, H.L.; et al. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 2015, 347, 1113–1117.
- Qin, L.; Kufareva, I.; Holden, L.G.; Wang, C.; Zheng, Y.; Zhao, C.; Fenalti, G.; Wu, H.; Han, G.W.; Cherezov, V.; et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015, 347, 1117–1122.
- Zheng, Y.; Han, G.W.; Abagyan, R.; Wu, B.; Stevens, R.C.; Cherezov, V.; Kufareva, I.; Handel, T.M. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 2017, 46, 1005–1017.e5.
- Pedragosa-Badia, X.; Stichel, J.; Beck-Sickinger, A.G. Neuropeptide Y receptors: How to get subtype selectivity. Front. Endocrinol. (Lausanne) 2013, 4, 5.
- Joedicke, L.; Mao, J.; Kuenze, G.; Reinhart, C.; Kalavacherla, T.; Jonker, H.R.A.; Richter, C.; Schwalbe, H.; Meiler, J.; Preu, J.; et al. The molecular basis of subtype selectivity of human kinin G-protein-coupled receptors. Nat. Chem. Biol. 2018, 14, 284–290.
- Eisen, H.N.; Hou, X.H.; Shen, C.; Wang, K.; Tanguturi, V.K.; Smith, C.; Kozyrytska, K.; Nambiar, L.; McKinley, C.A.; Chen, J.; et al. Promiscuous binding of extracellular peptides to cell surface class I MHC protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4580–4585.
- Ngo, T.; Ilatovskiy, A.V.; Stewart, A.G.; Coleman, J.L.; McRobb, F.M.; Riek, R.P.; Graham, R.M.; Abagyan, R.; Kufareva, I.; Smith, N.J. Orphan receptor ligand discovery by pickpocketing pharmacological neighbors. Nat. Chem. Biol. 2017, 13, 235–242.
- Michino, M.; Beuming, T.; Donthamsetti, P.; Newman, A.H.; Javitch, J.A.; Shi, L. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev. 2015, 67, 198–213.
- Kooistra, A.J.; Kuhne, S.; de Esch, I.J.; Leurs, R.; de Graaf, C. A structural chemogenomics analysis of aminergic GPCRs: Lessons for histamine receptor ligand design. Br. J. Pharmacol. 2013, 170, 101–126.
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071.
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265.
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095.
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217.
- Wu, F.; Song, G.; de Graaf, C.; Stevens, R.C. Structure and Function of Peptide-Binding G Protein-Coupled Receptors. J. Mol. Biol. 2017, 429, 2726–2745.
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the delta-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404.
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332.
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326.
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399.
- Zhang, C.; Srinivasan, Y.; Arlow, D.H.; Fung, J.J.; Palmer, D.; Zheng, Y.; Green, H.F.; Pandey, A.; Dror, R.O.; Shaw, D.E.; et al. High-resolution crystal structure of human protease-activated receptor 1. Nature 2012, 492, 387–392.
- Zheng, Y.; Qin, L.; Zacarias, N.V.; de Vries, H.; Han, G.W.; Gustavsson, M.; Dabros, M.; Zhao, C.; Cherney, R.J.; Carter, P.; et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540, 458–461.
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390.
- Oswald, C.; Rappas, M.; Kean, J.; Dore, A.S.; Errey, J.C.; Bennett, K.; Deflorian, F.; Christopher, J.A.; Jazayeri, A.; Mason, J.S.; et al. Intracellular allosteric antagonism of the CCR9 receptor. Nature 2016, 540, 462–465.
- Yin, J.; Babaoglu, K.; Brautigam, C.A.; Clark, L.; Shao, Z.; Scheuermann, T.H.; Harrell, C.M.; Gotter, A.L.; Roecker, A.J.; Winrow, C.J.; et al. Structure and ligand-binding mechanism of the human OX1 and OX2 orexin receptors. Nat. Struct. Mol. Biol. 2016, 23, 293–299.
- Yin, J.; Mobarec, J.C.; Kolb, P.; Rosenbaum, D.M. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 2015, 519, 247–250.
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139.
- Asada, H.; Horita, S.; Hirata, K.; Shiroishi, M.; Shiimura, Y.; Iwanari, H.; Hamakubo, T.; Shimamura, T.; Nomura, N.; Kusano-Arai, O.; et al. Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 2018, 25, 570–576.
- Cheng, R.K.Y.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.M.; Dumelin, C.E.; Dore, A.S.; et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017, 545, 112–115.
- Shihoya, W.; Nishizawa, T.; Okuta, A.; Tani, K.; Dohmae, N.; Fujiyoshi, Y.; Nureki, O.; Doi, T. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 2016, 537, 363–368.
- Yang, Z.; Han, S.; Keller, M.; Kaiser, A.; Bender, B.J.; Bosse, M.; Burkert, K.; Kogler, L.M.; Wifling, D.; Bernhardt, G.; et al. Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor. Nature 2018, 556, 520–524.
- Yin, J.; Chapman, K.; Clark, L.D.; Shao, Z.; Borek, D.; Xu, Q.; Wang, J.; Rosenbaum, D.M. Crystal structure of the human NK1 tachykinin receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 13264–13269.
- Robertson, N.; Rappas, M.; Dore, A.S.; Brown, J.; Bottegoni, G.; Koglin, M.; Cansfield, J.; Jazayeri, A.; Cooke, R.M.; Marshall, F.H. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 2018, 553, 111–114.
- Tanaka, H.; Yoshida, T.; Miyamoto, N.; Motoike, T.; Kurosu, H.; Shibata, K.; Yamanaka, A.; Williams, S.C.; Richardson, J.A.; Tsujino, N.; et al. Characterization of a family of endogenous neuropeptide ligands for the G protein-coupled receptors GPR7 and GPR8. Proc. Natl. Acad. Sci. USA 2003, 100, 6251–6256.
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019, 565, 318–323.
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the micro-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552.
- Liu, H.; Kim, H.R.; Deepak, R.; Wang, L.; Chung, K.Y.; Fan, H.; Wei, Z.; Zhang, C. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 2018, 25, 472–481.
- Krumm, B.E.; Grisshammer, R. Peptide ligand recognition by G protein-coupled receptors. Front. Pharmacol. 2015, 6, 48.
- Tikhonova, I.G.; Gigoux, V.; Fourmy, D. Understanding peptide binding in Class A G protein-coupled receptors. Mol. Pharmacol. 2019, 96, 550–561.
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376.
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844.
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048.
- Woolley, M.J.; Conner, A.C. Understanding the common themes and diverse roles of the second extracellular loop (ECL2) of the GPCR super-family. Mol. Cell. Endocrinol. 2017, 449, 3–11.
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.-P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers-aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31.
- Conway, P.; Tyka, M.D.; DiMaio, F.; Konerding, D.E.; Baker, D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014, 23, 47–55.
- Smith, C.A.; Kortemme, T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol. 2008, 380, 742–756.
- Bender, B.J.; Vortmeier, G.; Ernicke, S.; Bosse, M.; Kaiser, A.; Els-Heindl, S.; Krug, U.; Beck-Sickinger, A.; Meiler, J.; Huster, D. Structural Model of Ghrelin Bound to its G Protein-Coupled Receptor. Structure 2019, 27, 537–544.
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock ab-initio: Simultaneous folding, docking and refinement of peptides onto their receptors. PLoS ONE 2011, 6, e18934.
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814.
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 20–41.
More