Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Tomasz Ratajczyk and Version 2 by Lindsay Dong.
Since the discovery of triptycenes, great progress has been made regarding their synthetic methodology and the understanding of inter- and intramolecular interactions that involve triptycenes. Several new synthetic approaches have been developed in the last few years, and progress has been made in the context of sterically congested triptycenes and regioselective synthesis of various derivatives.
- triptycene
- synthesis
1. Introduction
The parent triptycene (9,10-dihydro-9,10[1′,2′]-benzenoanthracene) is an eye-catching hydrocarbon with a paddlewheel or propeller-shaped molecule. The characteristic structure of triptycene is composed of three benzene rings joined by two sp3 carbon atoms in a D3h-symmetric structure with a barrelene core (Figure 1).
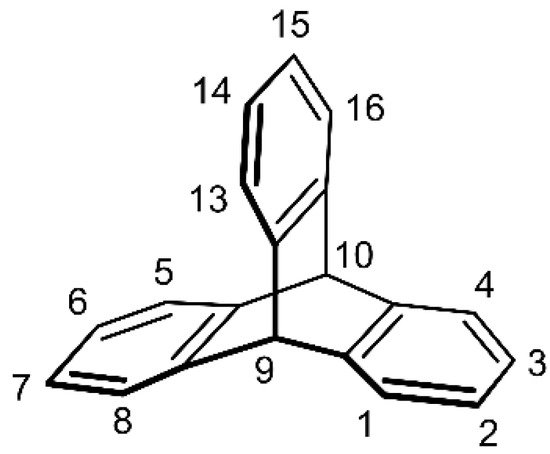
Figure 1. The chemical structure of the parent triptycene with a commonly used carbon atom numbering scheme based on the 9,10-dihydro-9,10[1′,2′]-benzenoanthracene nomenclature (CAS).
Triptycene was synthesized for the first time in 1942 by Bartlett et al., who also coined the name [1]. Fourteen years later, the rather laborious procedure became superseded by the more direct benzyne to anthracene cycloaddition route [2]. Since then, great progress has been made in the area of triptycenes synthesis, and recent developments are rich in examples of advanced structures, such as congested and structurally complex molecules [3][4][5][6][7][3,4,5,6,7].
Due to these inherent characteristics, triptycene has become an exemplar of a multipurpose molecular scaffold [8][9][10][11][12][13][14][15][16][17][18][19][20][21][9,10,11,12,13,14,15,16,17,18,19,20,21,22]. Triptycenes were used as building blocks for mechanically interlocked molecules [22][23][24][25][26][27][28][29][30][31][32][23,24,25,26,27,28,29,30,31,32,33] and for self-assembly and molecular recognition in the field of supramolecular chemistry [7][33][34][35][36][37][38][39][40][41][7,34,35,36,37,38,39,40,41,42]; as achiral or chiral ligands for catalysis [42][43][44][45][46][47][48][49][43,44,45,46,47,48,49,50]; anti-cancer agents [50][51][52][53][51,52,53,54]; models of hindered rotation [54][55][56][57][58][55,56,57,58,59]; as well as in broadly defined materials sciences [59][60][61][60,61,62]; as structural units in various functional polymers and porous materials [62][63][64][65][66][67][68][63,64,65,66,67,68,69], such as molecular cages [17][69][70][71][72][73][18,70,71,72,73,74]; and metal–organic frameworks [74][75][76][77][78][79][75,76,77,78,79,80]; as well as in chemical sensors [80][81][82][83][84][85][81,82,83,84,85,86] and liquid crystals [86][87][88][89][90][91][87,88,89,90,91,92].
2. Progress in Triptycene Synthesis and Derivatization
2.1. General Synthesis of the Triptycene Unit
The parent triptycene was obtained for the first time in 1942 by Bartlett et al., who used anthracene and 1,4-benzoquinone as starting materials (Scheme 1a) [1]. The rationale for this rather laborious, low-yielding, multi-step synthesis was laid in the study of radicals that can be generated on Csp3 bridgehead carbon atoms of triptycene (positions 9 and 10, Figure 1) [1][5][1,5].
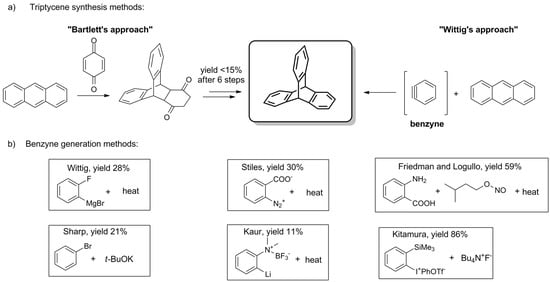
Scheme 1. (a) The general synthetic pathways for the synthesis of triptycenes [1][2][1,2]. (b) Benzyne generation methods: Wittig [2], Stiles [92][95], Friedman and Logullo [93][96], Sharp [94][97], Kaur [95][98], Kitamura [96][99].
Although the procedure was later simplified by Craig both in terms of time and steps required [97][94], a true breakthrough came with the distinct discovery of a cycloaddition reaction between anthracene and a direct synthon of triptycene’s benzene ring—benzyne. In 1956, Wittig and Ludwig were the first to report such a reaction of benzyne (Scheme 1a), being a reactive intermediate generated in situ from 2-fluorophenylmagnesium bromide (Scheme 1b) [2]. A few years later, Stiles described the synthesis of triptycene using o-diazonium benzoate (Scheme 1b) [92][95].
In 1963, Friedman and Logullo found anthranilic acid to be a superior benzyne precursor (Scheme 1b) [93][96]. During the reaction, anthranilic acid undergoes seamless diazotization by amyl nitrite. The resulting benzenediazonium-2-carboxylate fragmentates giving N2, CO2, and benzyne, which in turn engages in Diels–Alder reaction with anthracene. The feasibility of the procedure manifests itself in the good yield (59%) and the availability of starting materials. To this day, various benzyne precursors have been presented (Scheme 1b), some of which allowed for the considerable improvement of the yield [94][95][96][97,98,99].
2.2. Synthesis of Ortho-Functionalized Triptycenes
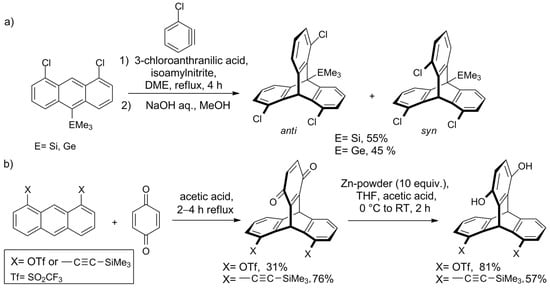
Electrophilic aromatic substitution (SEAr) reactions are disfavored at the ortho (α) positions respective to the bridgeheads. For example, nitration of the parent triptycene (HNO3/AcOH in Ac2O, 27–29 °C) yields products with the α to β substitution ratio of 1 to 40 [98][100]. This feature is a manifestation of the so-called “fused ortho effect” [99][101], according to which the reactivity toward electrophiles is largely diminished at the positions of the aryl ring that are ortho in respect to a fused strained ring [100][102]. In other words, a strained ring, such as triptycene’s bicyclo [2.2.2]octatriene, acts as a deactivating, meta/para directing group when fused with an arene. Since nitrotriptycenes provide access to the corresponding amines and other derivatives, such as alcohols and halogenated compounds, some optimized synthetic procedures for nitration were developed with regioselectivity in mind. Even though there are six β positions in the parent triptycene, mononitration to 2-nitrotriptycene was achieved with as high yield as 58–64% [79][101][80,103]. Optimized dinitration protocols yield 2,6-dinitrotriptycene (39%) [102][104], or a mixture of 2,6-/2,7-dinitrotriptycenes (56%) [103][105], and trinitration gives 2,7,14- and 2,6,14-trinitrotriptycene (21% and 64%, respectively) [104][106]. The potentially explosive product of an exhaustive nitration, 2,3,6,7,14,15-hexanitrotriptycene, was obtained in 42% yield [103][105]. Other SEAr reactions, such as acetylation [105][106][107,108] and formylation [106][108], also occur at β positions. Because of the fused ortho effect, α-functionalized triptycenes are usually prepared from appropriate prefunctionalized substrates. 1,8,13-Functionalized triptycenes epitomize an example of such an α-substitution pattern. The significance of this family originates from the structure–property relationship. The symmetry and rigidity of the triptycene itself, combined with the advantage of the substituents being located on the same face of the scaffold (pointing in one direction), facilitate the juxtaposition of functional groups in proximity. As tripodal molecules, 1,8,13-functionalized triptycenes recently found advanced application in surface modification—in 2015, Seiki et al. utilized the triptycene with three C12 side chains to self-assembly a remarkable organic thin film that was free of domain boundaries and structurally ordered up to the centimeter length scale [107][109]. The order and precise alignment of the molecular units of choice, which 1,8,13-functionalized triptycene allows for, are advantageous features for the promising field of high-performance organic thin-film materials [61][108][109][110][62,110,111,112]. The same 1,8,13 substitution pattern was used in models of molecular-level magnetic anisotropy [111][113], close nonbonded H–H contacts [112][114], and through-space Me–Ar interactions [113][115]. In 2010, the Mitzel group investigated the steric influence of the additional substituent at C-10 on the syn/anti ratio of 1,8,13/16-trichlorotriptycenes [114][117] and found that anti-isomer is generally favored. Unexpectedly, this was true for large substituents too, so the Mitzel group changed their focus from steric to electronic effects. Recently, they have investigated the cycloaddition of 3-chlorobenzyne to three 1,8-dichloroanthracenes equipped with CMe3, SiMe3, or GeMe3 group in position 10 [115][119]. It was observed that the electronic properties of the substituent influence regioselectivity—with electropositive SiMe3 and GeMe3, the syn triptycene formed predominantly, whereas for CMe3 only an anti-isomer was observed (Scheme 2a). Quantum chemistry calculations of the reaction pathway revealed that dispersion forces are one of the factors responsible for the observed effects.Scheme 2. Studies on the regioselectivity of the synthesis of ortho-substituted triptycenes by the Mitzel group: (a) the electronic character of a substituent on C10 influences the syn/anti ratio of the product [115][116][119,120]; (b) triptycenes functionalized with triflate or (trimethylsilyl)ethynyl groups [116][120].
Another approach aimed at regioselective synthesis of triptycene derivatives was presented by the group in 2018—Shwartzen et al. used cycloaddition of 1,4-benzoquinone to substituted anthracenes for the preparation of triptycenes functionalized with triflate or C≡C-SiMe3 groups (Scheme 2b)[116][120].
For the intermediate with two sterically demanding SiMe3 groups, a distortion of the molecular framework was observed. For that reason, attempts to obtain the 1,8,13-trisubstituted derivative by introduction of the third C≡C-SiMe3 were unsuccessful.
On the other hand, since the regioisomeric course of the Diels–Alder reaction depends on the nature of substituents in prefunctionalized substrates, approaches departing from chloroderivatives are also being explored. In their work reporting the synthesis of a porous molecular cube, Elbert at al. presented a path towards 1,8,13-trihydroxytriptycene [117][118], starting with the Diels–Alder reaction of 1,8-dimethoxyanthracene and 3-methoxybenzyne. The latter was generated in situ by a mild, fluoride-induced 1,2-elimination [118][121] of the corresponding trimethylsilyl triflate precursor [119][122]. A poorly soluble 2:1 mixture of syn- and anti-trimethoxytriptycenes was collected, and the target 1,8,13-trihydroxytriptycene was obtained after ether cleavage, esterification with capronyl chloride, crystallization of the desirable regioisomer, and hydrolysis of the pure 1,8,13-triester. The same method was used in the above-mentioned work of Seiki [107][109], who reported that syn-trihydroxytriptycene can be separated from the anti-isomer by recrystallization from DMF at −35 °C.
The challenge of sterically crowded triptycene synthesis was also taken by Szupiluk, who presented a route towards 9-nitro- and 9-aminotriptycenes (Scheme 3) [120][123]. The procedure comes down to cycloaddition of benzynes to 9-nitroanthracenes, followed by reduction of the nitro group. The reaction of electron-deficient anthracenes was not regioselective and accompanied by the formation of considerable amounts of side products. For example, 1,2,3,4-tetrabromo-9-nitrotriptycene was obtained in 7% yield; two other products were identified. On the other hand, it was demonstrated that the final reduction of nitro group on C9 in triptycenes proceeds smoothly and in a good yield, giving products equipped with the synthetically useful ammonium group.
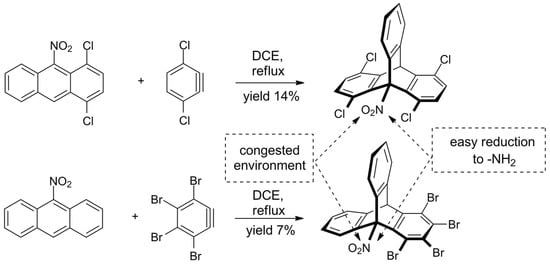
Scheme 3. Synthesis of new ortho-functionalized triptycenes from deactivated, electron-deficient nitroanthracenes [120][123].
Recently, Yoshinaga et al. published a follow-up article reporting some new developments in ynolate/benzyne chemistry [121][129]. The group presented the synthesis of 1,8,13-trisilylated triptycenes obtained from 3-silylbenzynes via the above-described triple cycloaddition route (Scheme 46).
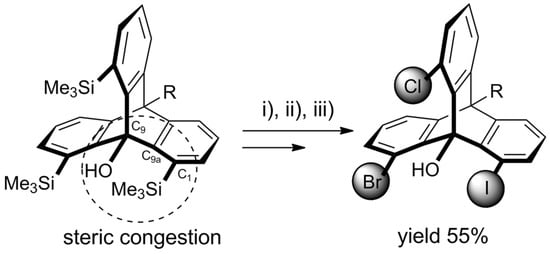
Scheme 46. Trisilyltriptycene (obtained according to Scheme 5 for R = Me and X = SiMe3) as a starting material for heterosubstituted triptycenes. An exemplary 1-bromo-8-chloro-13-iodo product was obtained with 55% overall yield in a chromatography-free procedure involving selective halogenations with (i) NCS (1.2 eq. in DMF, 60 °C, 2.5 h); (ii) NBS (2.0 eq. in DMF, 60 °C, 24 h); (iii) ICI (2.0 eq. in DCM, rt, 1 h) [121][129].
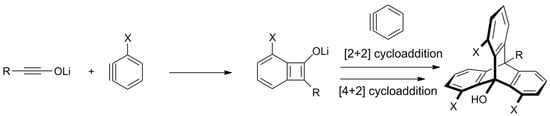
Scheme 5. A new approach towards substituted triptycenes—a regioselective synthesis from in situ generated ynolates and benzynes (X = H or OMe for R = Me, i-Pr, n-Bu, or Ph; X = t-Bu and R = H; X = n-Hex and R = OMe) [122].
2.3. Functionalization of the Bridgehead Positions of the Triptycene Unit
2.3. Functionalization of the Bridgehead Positions of the Triptycene Unit
In the chemistry of triptycene, the desired substitution pattern dictates the synthetic strategy. Low reactivity of triptycene’s aromatic ortho positions contrasts with the well-known activated character of the analogous position in simple alkyl-substituted arenes. In the same vein, while a benzylic position is generally reactive, the bridgehead C-H groups in triptycene are relatively inert to many reactions. Therefore, both ortho and 9,10-functionalized triptycenes are usually obtained from already functionalized precursors.
However, Oi et al. recently disclosed a Pd-catalyzed cross-coupling protocol that offers efficient functionalization of the bridgehead carbon atom in 9-bromotriptycene through C9-C bond formation (Scheme 67a) [123][131]. Various iodo- and bromoaromatic compounds were successfully tested as coupling partners, leading to a library of C9-Csp2 coupled products, which were obtained in good yields despite inherent steric hindrances. Illustrative C9-Csp coupling with (iodoethynyl)benzene and C9-Csp3 coupling with methyl bromoacetate were also successful. The procedure constitutes a highly attractive way of triptycene extension—its scope seems wide, the conditions are mild, and isolated yields were usually between 60% and 90%. Moreover, the required reagents—Pd(OAc)2 and tris(o-methoxyphenyl)phosphine (SPhos ligand)—are commercially available. 9-Triptycenylcopper, an active participant in the coupling reaction, forms in situ from 9-bromotriptycene treated with n-butyllithium followed by copper(I) halide (Scheme 67b). Additionally, the authors performed screening of other 9-metalated triptycene derivatives and found that 9-triptycenylboronate ester and 9-triptycenylzinc were not reactive under the tested conditions. In addition to triptycene, the scope of the work included C-C bond formation with other hindered structures—namely, adamantyl and mesityl.
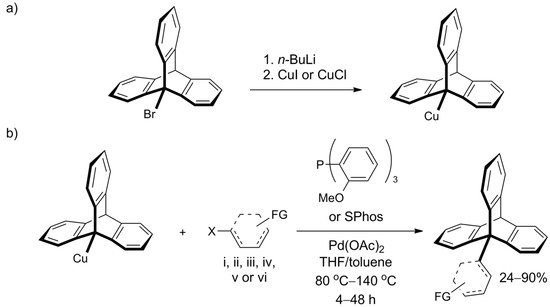
Scheme 67. An efficient coupling at position 9 of the triptycene, reported by Oi et al. [123][131]: (a) The preparation of the Cu derivative of triptycene (b) The coupling partner is composed of X = I or Br and an aromatic system (represented by a dashed structure) such as: (i) phenyl; (ii) benzene ring with a functional group (FG): o-CO2Me, p-CO2Me, p-CF3, p-CN, p-Ac, o- or p-OMe; (iii) thiophenyl substituted with FG=Ac or CO2Et; (iv) pyridinyl; (v) quinolinyl; or (vi) ferrocene.
2.4. Synthesis of Chiral Triptycenes
In the context of the structure–property relationship, another advantageous feature of the triptycene scaffold is its prochirality [124][132]. Indeed, many triptycenes with appropriate substitution patterns are optically active. The bridgehead carbon atoms can be seen as prochirality centers that can become asymmetric upon differentiation of their four substituents. For example, both 1,5- and 2,6-disubstituted triptycenes can, in principle, exist as two non-superimposable mirror images. The same holds true for 1,8- and 2,7-disubstituted triptycenes, provided that the substituents are different. Owing to the rigidity of the triptycene scaffold, the inherent chirality of such derivatives is permanent—there is no “risk” of racemization or inversion of configuration during chemical transformations. In 2020, an interesting review on chiral triptycenes in supramolecular and materials chemistry was presented by Preda et al. [7]. It is envisioned that chiral triptycenes will find applications in the growing field of circularly polarized luminescence (CPL) as CPL-active materials [125][126][127][128][129][130][133,134,135,136,137,138].
Sonoda reported enantiopure triptycenes obtained by recrystallisation of diastereomeric salts as early as in the 1960 s [131][139]. Recently, Ikai described a triptycene-based stationary phase for chiral HPLC [132][140]. The phase was effective in the resolution of axially chiral biaryls. The synthetic pathway began with a valuable racemic precursor, 2,6-dinitrotriptycene, obtained according to one of the above cited, optimized procedures [102][104].
In 2015, the first enantioselective synthesis of chiral triptycene was reported by Shibata [133][141]. Another one was recently presented by Aida [134][142], who proposed a strategy that starts from the preparation of chiral multicyclic cyclohexadienes. A set of such compounds was obtained by a highly enantioselective [2 + 2 + 2] cycloaddition between 2,2′-di(prop-1-yn-1-yl)-1,1′-biphenyls and 1,2-dihydronaphthalenes (Scheme 78). The reaction was catalyzed by a complex of rhodium(I) with a chiral (R)-SEGPHOS ligand, and the resulting cyclohexadienes were obtained with high enantiomeric excess. Subsequent Diels–Alder reaction with 1,4-naphthoquinone followed by reductive aromatization gave triptycene precursors. The final oxidative aromatization step was expected to convert the precursors to target triptycenes, but it turned out successful only in one of the cases. Nevertheless, a π-extended chiral triptycene with all three aromatic rings being different was obtained (87% ee).
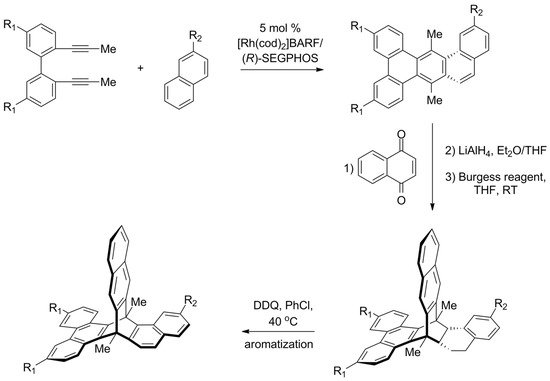
Scheme 78. Aida’s enantioselective synthesis of π-extended triptycene precursors, in which all three “paddles” are different (R1 = H or CF3, R2 = H or OMe) [134][142]. The last aromatization step was successful only for CF3/OMe combination of substituents.
To explain differences in the behavior of precursors, the authors investigated mechanistic aspects of the carbocation-involving oxidative dehydrogenation. It was determined that high strain causes an undesired carbocation rearrangement, which could be suppressed by the use of the electron-rich alkene and electron-deficient diyne.
2.5. Triptycenes in Catalysis
Triptycenes can be substituted with phosphorous, both on aromatic rings, as well as on bridgehead atoms. For example, over 20 years ago, Ramakrishnan presented 9-phosphinotriptycene [135][143] as a model for the study of the restricted rotation in the solid state (the compound was obtained from 9-lithiotriptycene and PCl3, followed by LiAlH4). Bearing in mind the rigid structure of the triptycene scaffold and the synthetic availability of its phoshine derivatives, an important branch of triptycene-related research emerged in recent years—the chemistry of triptycene-based phosphine ligands for catalysis. The area was pioneered by Gelman et al., both in the context of ligand synthesis, as well as their catalytic utilization [42][43][44][45][136][137][138][139][140][43,44,45,46,144,145,146,147,148].
In 2018, Kisets et al. presented a successful synthetic route towards 1,8,13-halogenated triptycenes and corresponding triphosphine ligands [141][149]. The method facilitates efficient synthesis of 1,8,13-tribromotriptycene derivative in two steps, starting with the Diels–Alder reaction of 1,8-dibromoanthracene towards 1,13-dibromo-6,7-dimethoxytriptycene (Scheme 89). Then, the intermediate was brominated giving a 1:1 mixture of syn- and anti-tribromotriptycenes that were separated by crystallization. Phosphination of the isolated syn-isomer yielded a pincer-like ligand with a rigidly arranged triphosphine coordination sphere. The reaction with PdCl2 resulted in carbometalation on Csp3-bridgehead atom. As evidenced by X-ray diffraction analysis, the geometry around the metallic center of this triptycene-based complex of Pd(II) was almost perfectly trigonal bipyramidal. It turned out that the compound is catalytically active during transfer hydrogenation of α,β-unsaturated ketones. The reaction is chemoselective owing to the singular steric environment around the catalytic site.
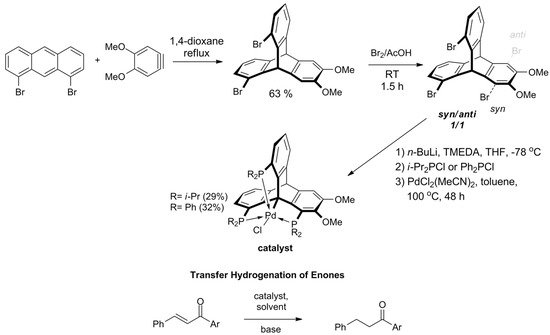
Scheme 89. The first applicable synthesis of 1,8,13-tribromotriptycene. Phosphination and bridgehead carbon atom metalation gives triptycene-based complexes of Pd(II), which act as catalysts for chemoselective transfer hydrogenation of α,β-unsaturated ketones (R = i-Pr; Ar = Ph, 4-MeO-phenyl, 1-naphthyl, 2-furyl) [141][149].
The methods for the synthesis of catalysts, combined with the chiral nature of many derivatives, open the prospect for the deployment of triptycenes in asymmetric synthesis. The first case of enantioinduction due to a chiral monophosphinotriptycene-containing catalyst was reported in 2018. Leung et al. [142][150] noted that while bisphosphine ligands based on 1,8-disubstituted triptycenes are well researched, monophosphinotriptycenes are no less interesting—in general, monodentate ligands are highly active in metal-catalyzed reactions. The authors efficiently prepared an exemplary ligand, 1-methoxy-8-(diphenylphosphino)triptycene, in five steps starting from 1,8-dihydroxytriptycene. By design, the desymmetrization of the molecule occurred upon P(O)Ph2 group introduction, and the enantiomers were separated by chiral chromatography of the penultimate intermediate. Next, the catalytic activity of a monophosphinotriptycene scaffold was demonstrated for the first time in tests against Pd-catalyzed Suzuki–Miyaura cross-coupling. Although the racemic ligand allowed for biaryls to be obtained in excellent yields (>90% in most cases, Pd(II) at 0.001 mol%) (Scheme 910a), no enantiomeric excess was observed in tests utilizing an optically active triptycene for the coupling of 2-methylphenylboronic acid with 1-bromo-2-methylnaphthalene or 1-bromo-2-methoxynaphthalene.
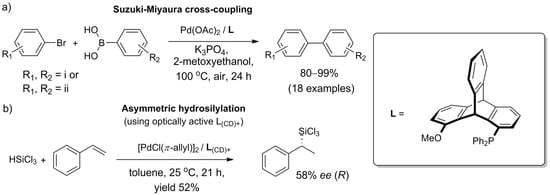
Scheme 9. Catalytic utility of triptycene-based monophosphine ligand obtained by Leung et al. [143]: (a) in Suzuki–Miyaura cross-coupling with racemic ligand, the scope was successfully tested with the following substituents: (i) R1 = o-Me, o-OMe, m-OMe, m-NH2, p-Me, p-OMe, p-COMe, p-CHO, p-NO2, or p-CF3, provided that R2 = H; and (ii) all combinations of R1 = p-NO2, p-COMe, or p-OMe with R2 = p-CF3, m,p-(OMe)2, or o-Me; (b) in asymmetric hydrosilylation of styrene, chiral ligand facilitated the formation of chiral product (58% ee, as determined for the corresponding alcohol obtained via Fleming–Tamao oxidation.
The catalytic uses of triptycene go beyond the sphere of phosphine ligands. Some interesting results were recently presented by Nishii et al. [144][151]. The authors reported that 9-methylsulfanyltriptycene (TRIP-SMe) catalyzes the halogenation of aromatic compounds, including unactivated ones, by N-halosuccinimides (NXS) (Scheme 101a). The approach offers a route towards haloarenes in which harsh reaction conditions and/or classical electrophilic halogenation using bromine or chlorine can be avoided. TRIP-SMe catalyst was easily prepared by treating 9-bromotriptycene with n-BuLi and dimethyldisulfide, followed by two-step purification by chromatography and recrystallization (84% yield). The synthetic procedure, which allowed for successful halogenation of various aromatic compounds under mild reaction conditions, combines the aromatic substrate, NXS, TRIP-SMe catalyst, and AgSbF6 activator (Scheme 110b). According to the proposed mechanism, TRIP-SMe captures halogen from NXS to become a halonium adduct, TRIP-S(Me)X+ (Scheme 110a).
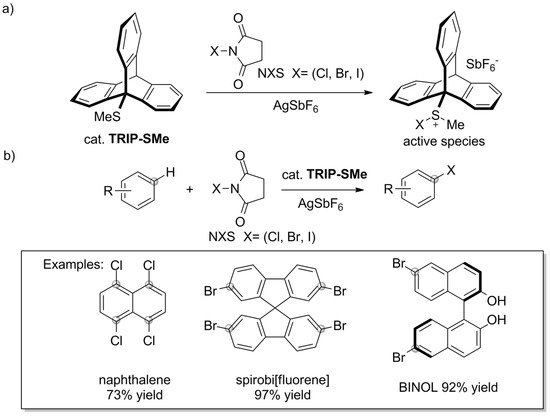
Scheme 101. (a) Methylsulfanyltriptycene/AgSbF6 catalyst system reported by Nishii et al. [144][151] and (b) the mild halogenation of aromatic compounds by N-halosuccinimides.
3. Conclusions
In conclusion, the advances in synthetic methodology and a better understanding of the relation between triptycene structures and their properties have been very significant. Moreover, the progress of triptycene synthesis and comprehensive insight into the nature of these interactions is of central concern for the rational design of triptycene-based functional materials. However, some questions remain open, and the chemistry of triptycenes and their applications will still be developing.