1. Autophagy
Cellular homeostasis is dependent on intracellular mechanisms that maintain organelles and functional macromolecules that are required for cell survival and normal biosynthetic function
[1]. Autophagy is an evolutionarily conserved homoeostatic process
[2], initially described by Deter et al. in 1960
[3], and is characterized by being a highly conserved process by which cytoplasmic components (cytosolic macromolecules and dysfunctional organelles) are generally marked by ubiquitination and delivered to lysosomes for degradation and recycling
[4][5][6][4,5,6]. This serves as a quality control mechanism as well as a recycling pathway
[2]. There are at least three types of autophagy in eukaryotic cells: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), differing between them in relation to the mode of delivery of the degradation contents to the lysosome
[7][8][9][7,8,9]. Autophagy has a controversial role because it plays a dual function in both cell survival and apoptosis, since it can be used as a protection and a death mechanism in stressed cells
[10][11][12][13][10,11,12,13], and it is suggested that the type of response depends on the cellular context
[10][11][10,11]. Of all the known forms of cell death, including apoptosis, necrosis, and pyroptosis, among others
[13], autophagy is described as a programmed non-apoptotic cell death
[14]. The role of autophagy in facilitating cell death is very important, since it can remove senescent cells in aging tissues in addition to controlling the growth of neoplastic lesions
[13].
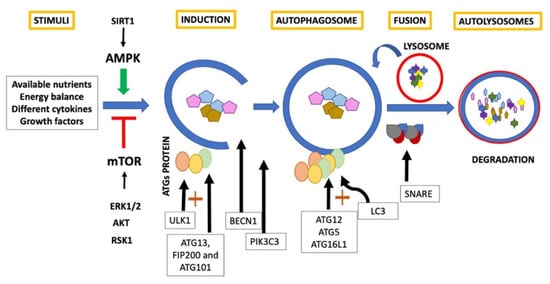
In general, classic autophagy consists of a series of dynamic membrane arrangements mediated by a group of Autophagy Related Proteins (ATG)
[1][6][1,6]. First, cytoplasm sequestration is generated within double-membrane vesicles called autophagosomes
[5][6][9][5,6,9]. ULK1 is an autophagy inducer
[9][10][11][9,10,11], and forms the first complex of this pathway with ATG13, FIP200 and ATG101
[2]. Beclin1 (BECN1), acts as an autophagy regulator
[9][10][11][9,10,11] and, in conjunction with phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3) and phosphoinositide-3-kinase regulatory subunit 4 (PI3KR4), promotes engulfment and endosome maturation
[15]. ULK complex is mobilized to the endoplasmic reticulum favoring the formation of the phagophore and allowing the recruitment of WIPI2B that recruits the E3-like complex ATG12– ATG5–ATG16L1
[16]. This last complex promotes the conjugation of LC3
[17]. LC3 is a critical biomarker of autophagosome formation and expansion and, when conjugated with phosphatidylethanolamine, results in the formation of another autophagy marker LC3-II
[9][10][11][9,10,11]. Subsequently, the autophagosome fuses with the lysosome to generate autolysosomes guided by SNARE proteins
[16]. Finally, the intracellular components are rapidly degraded by autophagolysosomal hydrolytic enzymes
[17] and the constituents are released for biosynthesis or for their use as an energy source
[5][6][9][5,6,9] (
Figure 1).
Figure 1. Classic autophagy.
Autophagy pathways are integrated into multiple-signal transduction pathways that respond to the amount of available nutrients, energy balance, different cytokines, and growth factors
[9]. A vital autophagy regulator is the interaction between serine and threonine kinases of the mammalian target of rapamycin (mTOR) in the mTOR 1 complex (mTORC-1)
[18]. It functions as a central regulatory protein that integrates signals originating from intracellular and extracellular changes
[19]. Inhibition of mTORC-1 promotes autophagy and activation of mTOR kinase suppresses autophagy
[6][20][6,20]. Activation of extracellular signal-regulated kinase 1/2 (ERK1/2), protein kinase B (AKT), and p90 ribosomal S6 kinase (RSK1) promotes the activity of mTORC1
[19]. In fact, ERK1/2 signal cascade is activated in several autophagy models, and inhibition of this pathway inhibits autophagy
[21]. Adenylate-activated protein kinase (AMPK) is a serine/threonine protein kinase that is activated through a combination of multiple phosphorylation by upstream kinases. AMPK is able to promote autophagy by acting differentially at different levels of autophagy regulation, for example by inhibition of mTORC-1 and phosphorylation of ULK1
[17][22][17,22]. Pathways of mTOR and AMPK are also linked to Sirtuin 1 (SIRT1)
[23]. SIRT1 is a member of the class III histone deacetylase (HDAC) family and interacts with proteins linked to the regulation of autophagy such as ATG5, ATG7, LC3, Forkhead box O transcription factors (FoxO), E2F transcription factor 1 (E2F1), tumor protein p73 (TP73), PPAR-γ co-activator 1α (PGC1α), NF-κB and tumor protein p53 (TP53)
[23]. Overexpression and activation of SIRT1 by resveratrol induces protective autophagy in non-small-cell lung cancer cells (NSCLC) via inhibiting Akt/mTOR and activating p38-MAPK pathway
[24]. Even this activation of SIRT1 has been shown to be capable of inducing autophagy in enucleated cells
[6]. Further, the reduction in the availability of nutrients generates the activation of sensors such as SIRT1, AMPK and mTOR, which increases the demand for autophagic replacement
[4]. Finally, nuclear factor κB (NF-κB) is a transcriptional factor and is an important inducer of several autophagy related genes such as BECN1, BCL2 and sequestosome 1 (SQSTM1)
[25]. It has been observed in degenerative human nucleus pulposus cells that inhibition of NF-κB blocks apoptosis and inflammation by promoting autophagy through AKT/mTOR pathway
[26].
2. Autophagy and Aging
Autophagy is generating great interest due to its role in several physiological processes that are important for health and in age-related degenerative diseases
[10][27][10,27].
Aging is frequently accompanied by defects in general autophagy, which causes a decrease in the ability of organisms to adapt to stress
[5] and the accumulation of metabolic wastes, typical of cellular aging
[28]. These effects have been observed in different aged rat tissues such as kidney, heart, cartilage, brain, and skeletal muscle, in which a significant decrease in the expression of LC3, BECN1 and ULK1 has been reported
[29]. This decline in autophagic capacity in aging cells alters the cell maintenance process, promoting ROS generation and oxidative stress
[22]. ROS accumulation and mTORC1 activation, are associated with accelerated aging and the development of age-related pathologies
[1]. Some of these age-related diseases are characterized by the accumulation of autophagic vacuoles, which supports that the autophagic process is progressively altered with aging
[30]. Finally, defects in autophagy can aggravate age-related alterations in model organisms, while the activation of autophagy protects against diseases related to aging and could lengthen life and reduce the severity of the disease
[9][25][9,25].
In cartilage, autophagy is also considered a protective mechanism, for maintaining homeostasis and for being a cellular response to different types of stress
[9][10][26][30][9,10,26,30]. It is thought that the development of structural changes in cartilage due to aging is linked to the alteration in homeostatic mechanisms such as autophagy
[25][28][25,28] and that defects in autophagy cause tissue degeneration similar to that associated with aging
[9][25][9,25]. It has been postulated that one of the connections could be the FoxO protein, given that FoxO controls: chondrocyte proliferation, cell viability by coordinating key cellular stress responses, articular cartilage homeostasis during aging and its overexpression significantly increased autophagic genes
[31].
3. Autophagy and Osteoarthritis
Osteoarthritis (OA) is a complex and multifactorial degenerative joint disease, it is one of the main causes of pain and dysfunction worldwide
[29][32][33][34][29,32,33,34]. It is characterized by a degradation of the articular cartilage and a concomitant adaptive osteogenesis
[30][35][30,35]. Although the cartilage has the most notable changes, the entire joint is affected, including the synovium, joint ligaments, and subchondral bone and it has been observed that inflammation from both synovitis and systemic inflammation play an important role in the genesis of this disease
[31][36][31,36]. The chondrocyte itself contributes to joint degradation through enzymatic degradation of the extracellular matrix (ECM) (chondrocytic chondrolysis)
[35]. Some of the changes that can be observed are: vascular infiltration, osteophyte formation, activation of macrophage, hypertrophic chondrocytes, fibrotic synovium, sclerotic bone formation, etc.
[37].
Despite the fact that aging is one of the most important risk factors for OA
[26][29][31][32][33][34][38][26,29,31,32,33,34,38], osteoarthritis is probably not a direct consequence of this, but it is aging itself that affects the ability of articular cartilage to maintain homeostasis such as autophagy
[39]. Both aged human and mouse cartilage shows a reduction in autophagic protein expression
[33]. By observing the formation of autophagic vesicles in cartilage as a measure of autophagic activity, Caramés et al. (2015) could demonstrate that there is a significant reduction in the level of basal autophagy in aged mice compared to young mice, and also mention that with aging there is a decrease in the expression of ATG5 and LC3 and that structural damage progresses in an age-dependent manner subsequent to changes in autophagy expression
[33]. Also a reduction and loss of ULK1, BECN1, and LC3 has been associated with an increase in chondrocyte apoptosis in OA
[1][11][1,11]. The defects in autophagy regulation in chondrocytes and aged cartilage have also been observed in OA models
[40].
In OA a functional relationship between autophagy and apoptosis is also described, where in early stages of OA autophagy would be active to protect chondrocytes
[14][41][14,41], while in later stages, autophagy could be active together with the apoptosis as an alternative pathway to cellular demise and could even induce senescence
[14][42][14,42]. In this sense, it has been indicated that in chondrocytes and cartilage with OA versus healthy patients, autophagy may be increased with increased expression of autophagic markers. This increase is thought to be an adaptive response to protect cells from stress and regulate changes in OA-related gene expression through modulation of apoptosis and ROS during the phase initial degenerative OA, but in cases of severe damage autophagy would decrease
[37][43][37,43]. In addition, it has been observed that the inflammatory stimulus with IL-1β in chondrocytes increases the LC3I protein expression similar to the one that was observed with the autophagy inducer, rapamycin, and this excessive or prolonged activation could promote cell death
[44], these results are consistent with other studies
[39][40][41][39,40,41].
On the other hand, it has been described that deletion of autophagy proteins could influence the development of OA. KO mice for some FoxO proteins generate alterations similar to those observed in cartilage due to OA and the overexpression of Fox1 reduces inflammatory mediators in chondrocytes with OA
[31]. Since mTOR has been described to be overexpressed in human cartilage with OA, its deletion has been reported to favor protection against destabilization of medial meniscus (DMM)-induced OA
[45].
There is no current cure for OA. Treatment can be classified into reduction of modifiable risk factors, intra-articular therapy, physical modalities, alternative therapies, and surgical treatments
[46]. Exercise, patient education, and weight loss are the first line treatment recommended for knee and hip OA
[47].
In the pharmacological aspect, there are no drugs available that are capable of modifying OA and there are a large number of drug candidates in clinical trials that have failed to demonstrate efficacy or are associated with adverse effects
[1]. The most used pharmacological treatments in OA are generally symptomatic and focus on non-steroidal anti-inflammatory drugs (NSAIDs) and analgesics
[44][48][44,48] and COX-2 inhibitors such as rofecoxib, that decrease pain but cannot stop disease progression, and intra-articular glucocorticoid and hyaluronic acid injections that decrease pain but may also increase disease progression
[49]. For this reason, other therapeutic alternatives are necessary to prevent or decrease the progression of OA, avoiding the side effects of most treatments and focusing on the multimodal and progressive nature of OA
[50].
4. Autophagy as a Therapeutic Target in OA
There are currently several treatments for OA that promote autophagy. An example is rapamycin, an mTOR inhibitor, which induces autophagy in various cell types including in particular chondrocytes limiting joint damage
[9][51][9,51]. Autophagy activation by rapamycin reduces intracellular levels of ROS induced by IL-1β and reduces cartilage destruction in experimental OA models
[41]. Rapamycin is a useful drug for the study of OA in different models, but it can have potential adverse effects, such as disorders of the blood, metabolism, and nervous system, among others, which limits its use in long-term treatments
[9]. Another drug that is safer for long-term administration and is commonly prescribed to relieve OA in humans is glucosamine
[52]. Glucosamine prevents demethylation of IL-1β resulting in decreased expression
[53], it also modulates targets of the autophagy pathway in vitro and in vivo in a manner dependent on the AKT/FoxO/mTOR pathway
[9]. Some researchers have investigated other ways of regulating autophagy, for example through microRNAs. The miRNA-335-5p activates autophagy in human OA chondrocytes, increasing its viability and reducing its inflammatory mediators
[54]. On the other hand, deregulation of miR-128a targeting ATG12, impairs chondrocyte autophagy and accelerates development of OA, and its interruption attenuated chondrocyte dysfunction and delayed OA development
[55]. The development of safe and effective drugs that can enhance autophagic activity or restore autophagy flux is a promising strategy for the treatment of OA
[42].
There are several components that have been suggested to have anti-senescence activity, among these are polyphenols, especially for their antioxidant and anti-inflammatory activity at the systemic level
[45][56][45,56].