This entry describes a drug repurposing study that identifies entrectinib as a potential antiviral drug against SARS-CoV-2.
This entry describes entrectinib as an antiviral drug
- SARS-CoV-2
- COVID-19
1. Background
2. Targeting the RBD–ACE2 Interface for a SARS-CoV-2-Specific Antiviral Action
A promising strategy to prevent SARS-CoV-2 infections is to inhibit the initial step of viral entry into the host cell. The SARS-CoV-2 spike protein and in particular the receptor-binding domain (RBD) are crucial for viral attachment to the ACE2 host cell receptor.
2.1. Molecular Dynamics Simulations of the RBD–ACE2 Interface Reveal Contact Hotspots
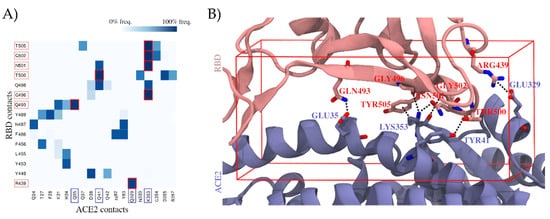
2.2. Virtual Screening Yields Two Drug Candidates with the Potential to Inhibit SARS-CoV-2 Cell Entry
| Compound Name | Source | Docking Assay | |
|---|---|---|---|
| Nilotinib | Virtual screening | RBD docking | |
| Entrectinib | Virtual screening | ACE2 docking | |
| Rutin | Virtual screening | ACE2 docking | |
| Diosmin | Virtual screening | ACE2 docking | |
| Naldemedine | Virtual screening | RBD and ACE2 docking | |
| Apilimod | Literature [18] (used as positive control in our validation experiments) | Literature [21] (used as positive control in our validation experiments) |
- |
| Argatroban | Literature [20] | Literature [19] | - |
| Otamixaban | Literature [20] | Literature [19] | - |
| Ivermectin | Literature [19] | Literature [18] | - |
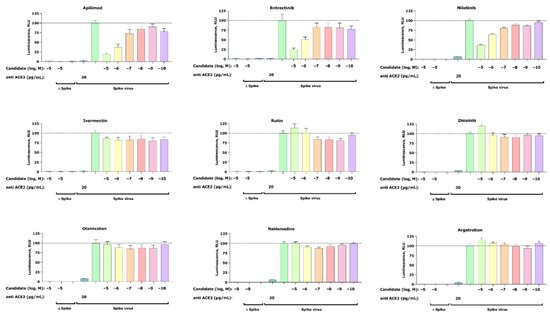
2.3. Proof of Concept
2.3.1. Pseudovirus Assay Confirms Anti-SARS-CoV-2 Activity for Entrectinib and Nilotinib
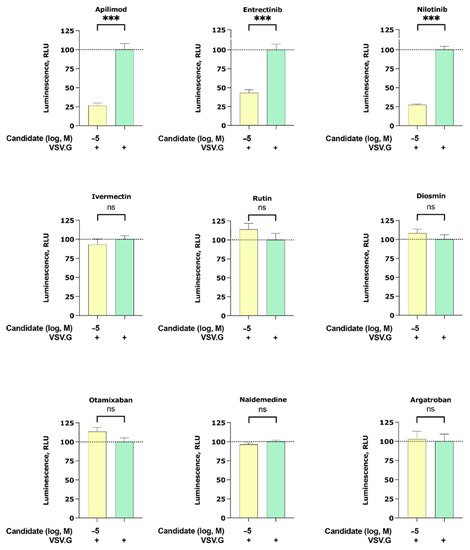
2.3.2. Apilimod, Entrectinib, and Nilotinib Inhibit VSV.G Cell Entry
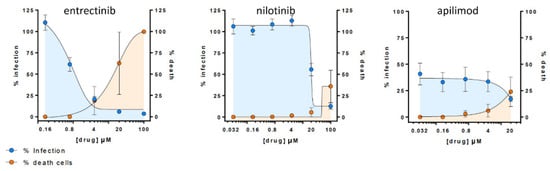
2.3.4. Entrectinib Inhibits Cell Infection in Human Lung Tissue (HLT) Cells at Non-Cytotoxic Concentrations
3. Conclusion
OurThe study reports antiviral activity of entrectinib against SARS-CoV-2 in human lung tissue, which has not been previously reported. Entrectinib is an FDA-approved drug for the treatment of solid tumors with NTRK fusion proteins and for ROS1-positive non-small cell lung cancers. Ultimately, further studies are required to confirm these data and the molecular mechanism of action for entrectinib.
References
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 29 November 2021).
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic Characterization of the 2019 Novel Human-Pathogenic Coronavirus Isolated from a Patient with Atypical Pneumonia after Visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236.
- Cheng, V.C.C.; Lau, S.K.P.; Woo, P.C.Y.; Yuen, K.Y. Severe Acute Respiratory Syndrome Coronavirus as an Agent of Emerging and Reemerging Infection. Clin. Microbiol. Rev. 2007, 20, 660–694.
- Cui, J.; Li, F.; Shi, Z.-L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192.
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569.
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.-Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a PH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains. Cell Host Microbe 2020, 28, 867–879.
- Alshammary, A.F.; Al-Sulaiman, A.M. The Journey of SARS-CoV-2 in Human Hosts: A Review of Immune Responses, Immunosuppression, and Their Consequences. Virulence 2021, 12, 1771–1794.
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826.
- Lu, H. Drug Treatment Options for the 2019-New Coronavirus (2019-NCoV). Biosci. Trends 2020, 14, 69–71.
- Pavan, M.; Bolcato, G.; Bassani, D.; Sturlese, M.; Moro, S. Supervised Molecular Dynamics (SuMD) Insights into the Mechanism of Action of SARS-CoV-2 Main Protease Inhibitor PF-07321332. J. Enzyme Inhib. Med. Chem. 2021, 36, 1646–1650.
- Yavuz, S.; Komsuoğlu Çelikyurt, F.I. Antiviral Treatment of COVID-19: An Update. Turk. J. Med. Sci. 2021.
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin Has Potent Preclinical Efficacy against SARS-CoV-2 by Targeting the Host Protein EEF1A. Science 2021, 371, 926–931.
- Falcone, M.; Tiseo, G.; Valoriani, B.; Barbieri, C.; Occhineri, S.; Mazzetti, P.; Vatteroni, M.L.; Suardi, L.R.; Riccardi, N.; Pistello, M.; et al. Efficacy of Bamlanivimab/Etesevimab and Casirivimab/Imdevimab in Preventing Progression to Severe COVID-19 and Role of Variants of Concern. Infect. Dis. Ther. 2021, 10, 2479–2488.
- Blaess, M.; Kaiser, L.; Sommerfeld, O.; Csuk, R.; Deigner, H.-P. Drugs, Metabolites, and Lung Accumulating Small Lysosomotropic Molecules: Multiple Targeting Impedes SARS-CoV-2 Infection and Progress to COVID-19. Int. J. Mol. Sci. 2021, 22, 1797.
- Hoertel, N.; Sánchez-Rico, M.; Gulbins, E.; Kornhuber, J.; Carpinteiro, A.; Lenze, E.J.; Reiersen, A.M.; Abellán, M.; de la Muela, P.; Vernet, R.; et al. Association Between FIASMAs and Reduced Risk of Intubation or Death in Individuals Hospitalized for Severe COVID-19: An Observational Multicenter Study. Clin. Pharmacol. Ther. 2021, 110, 1498–1511.
- Carpinteiro, A.; Edwards, M.J.; Hoffmann, M.; Kochs, G.; Gripp, B.; Weigang, S.; Adams, C.; Carpinteiro, E.; Gulbins, A.; Keitsch, S.; et al. Pharmacological Inhibition of Acid Sphingomyelinase Prevents Uptake of SARS-CoV-2 by Epithelial Cells. Cell Rep. Med. 2020, 1, 100142.
- Hoertel, N.; Sánchez-Rico, M.; Vernet, R.; Beeker, N.; Jannot, A.-S.; Neuraz, A.; Salamanca, E.; Paris, N.; Daniel, C.; Gramfort, A.; et al. Association between Antidepressant Use and Reduced Risk of Intubation or Death in Hospitalized Patients with COVID-19: Results from an Observational Study. Mol. Psychiatry 2021, 26, 5199–5212.
- Lehrer, S.; Rheinstein, P.H. Ivermectin Docks to the SARS-CoV-2 Spike Receptor-Binding Domain Attached to ACE2. In Vivo 2020, 34, 3023–3026.
- Rensi, S.; Altman, R.B.; Liu, T.; Lo, Y.-C.; McInnes, G.; Derry, A.; Keys, A. Homology Modeling of TMPRSS2 Yields Candidate Drugs That May Inhibit Entry of SARS-CoV-2 into Human Cells. ChemRxiv 2020.
- Gayle, S.; Landrette, S.; Beeharry, N.; Conrad, C.; Hernandez, M.; Beckett, P.; Ferguson, S.M.; Mandelkern, T.; Zheng, M.; Xu, T.; et al. Identification of Apilimod as a First-in-Class PIKfyve Kinase Inhibitor for Treatment of B-Cell Non-Hodgkin Lymphoma. Blood 2017, 129, 1768–1778.
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 Antiviral Drugs through Large-Scale Compound Repurposing. Nature 2020, 586, 113–119.
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620.
- Baranov, M.V.; Bianchi, F.; van den Bogaart, G. The PIKfyve Inhibitor Apilimod: A Double-Edged Sword against COVID-19. Cells 2021, 10, 30.
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.
- Grau-Expósito, J.; Perea, D.; Suppi, M.; Massana, N.; Vergara, A.; Soler, M.J.; Trinite, B.; Blanco, J.; García-Pérez, J.; Alcamí, J.; et al. Evaluation of SARS-CoV-2 Entry, Inflammation and New Therapeutics in Human Lung Tissue Cells. bioRxiv 2021.