Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by John Karalis.
Colorectal cancer (CRC) is the third most common malignancy and the second most common cause of cancer-related mortality worldwide. A total of 20% of CRC patients present with distant metastases, most frequently to the liver and lung. In the primary tumor, as well as at each metastatic site, the cellular components of the tumor microenvironment (TME) contribute to tumor engraftment and metastasis. These include immune cells (macrophages, neutrophils, T lymphocytes, and dendritic cells) and stromal cells (cancer-associated fibroblasts and endothelial cells).
- tumor microenvironment
- colorectal cancer
- colorectal liver metastasis
- colorectal pulmonary metastasis
- immuno-oncology
- novel anticancer therapy
1. Introduction
Colorectal cancer (CRC) is the third most common malignancy and the second leading cause of cancer-related mortality worldwide [1]. Metastasis is a primary driver of CRC-related mortality, with the liver and lungs representing the most frequently involved organs. While surgical resection of colorectal liver metastases (CRLMs) and colorectal pulmonary metastases (CRPMs) provides the only potentially curative treatment, select patients with unresectable metastatic CRC (mCRC) may benefit from other locoregional therapies, including radiofrequency ablation and stereotactic radiotherapy [2,3][2][3]. However, for most patients, systemic chemotherapy is the cornerstone of treatment. Recently, immune checkpoint inhibitors (ICIs) have been shown to have a significant clinical benefit in CRC that has high microsatellite instability or mismatch repair deficiencies [4]. This advance reflects the potential of tumor microenvironment (TME)-directed therapies for the treatment of mCRC.
2. Colorectal Liver Metastasis (CRLM)
The liver is the most common site of CRC metastases [23,24][5][6]. Up to 25% CRC patients may have synchronous CRLM, with an additional 50% developing CLRM during their disease course [25][7]. Oncogenotype may predict CRLM; Chu et al. [26][8] demonstrated that mutant KRAS+ CRC (found in 30–40% of cases) is associated with CRLM through the upregulation of YB-1 and insulin-like growth factor-1 receptor via the MEK-miR137 signaling pathway. Consistently, Brudvik et al. [27][9] identified that mutant KRAS status was inversely associated with overall and recurrence-free survival after CRLM resection in a meta-analysis of 1809 patients. The liver microenvironment represents a unique metastatic niche, and there may be differences in the immune landscape between liver metastases, the primary tumor, and other metastatic sites. Indeed, Tian et al. [28][10] demonstrated that CRC cells cultured on decellularized liver and lung scaffolds formed spheroid “metastases” in vitro and exhibited organ-specific tropism when injected into murine-models. Wei et al. showed in a 74-patient cohort that programmed death-ligand 1 (PD-L1) expression, a potential biomarker for ICI efficacy, and CD4 T-cell density were higher in CRLMs than the primary tumor [29][11]. An improved understanding of the CRLM TME, therefore, has implications on treatment strategy and may justify biopsy excisional biopsy of a CRLM to analyze both the metastasis and its microenvironment.
2.1. Pathophysiology of Colorectal Liver Metastasis
CRCs are thought to seed the liver primarily through the portal venous circulation. In contrast, cancer cells originating from outside of the gastrointestinal tract likely seed through the systemic circulation via the hepatic artery. These two sources of hepatic blood flow join at the point of entry into the liver sinusoids, where blood perfuses the liver parenchyma and returns to the systemic circulation via the centrilobular veins. Once circulating CRC tumor cells enter the liver sinusoids, the seeding of metastasis to the liver occurs in four dynamic, overlapping phases (Figure 1) [30,31,32][12][13][14].
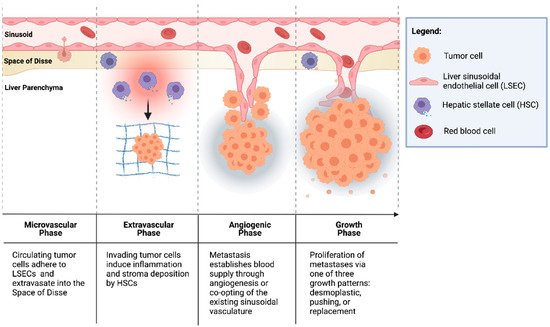
Figure 1. Formation of colorectal liver metastases through four overlapping phases: microvascular, extravascular, angiogenic, and growth.
2.1.1. Microvascular Phase
Once in the portal vascular system, CRC circulating tumor cells pass through the portal venules into the sinusoidal capillaries. There, the cells encounter liver-specific defense mechanisms, and many are phagocytosed by stellate macrophages (Kupffer cells (KCs)), a process potentiated by natural killer cells (NKs) [33,34][15][16]. However, cells that escape innate immune surveillance must stop in the sinusoids to extravasate and form a metastatic lesion. While this mechanical arrest contributes to the adhesion of circulating tumor cells to the sinusoidal vasculature, intravital microscopy has shown that CRC circulating cells are usually smaller than the vessel diameter [35,36][17][18]. This suggests that specific cell-adhesion interactions can facilitate tumor cell arrest.
The liver sinusoids are lined by liver sinusoidal endothelial cells (LSECs), which express the critical cell-adhesion protein, E-selectin. E-selectin is a cytokine-inducible protein implicated in CRC cell adhesion to liver sinusoidal endothelial cells [37][19]. Notably, the inhibition of endothelial E-selectin has been shown to reduce the number of CRLMs up to 97% relative to the placebo in syngeneic murine models [37,38][19][20]. Furthermore, Khatib et al. [39,40][21][22] showed that CRC circulating cells trigger the release of proinflammatory TNF-α and IL-1β from KCs, thereby upregulating the expression of E-selectin and other adhesion molecules on liver sinusoidal endothelial cells and enhancing liver metastases [39,40][21][22].
2.1.2. Extravascular/Pre-Angiogenic Phase
Once tumor cells have extravasated from the sinusoidal microvasculature into the space of Disse, hepatic stellate cells (HSCs) are activated by the pro-inflammatory cascade that was initiated in the microvascular phase. HSCs produce extracellular matrix (ECM) proteins, including collagen, laminin, fibrillin, and fibronectin, and provide a scaffold for tumor cells [41,42][23][24]. Additionally, KCs and neutrophils secrete matrix metalloproteinases (MMPs) and elastase, which degrade and remodel the ECM, facilitating tumor cell invasion [31,43][13][25]. Furthermore, cells in the TME have been implicated in the induction of an immunosuppressive niche, facilitating tumor cell growth. Huang et al. [44][26] demonstrated that HSCs co-cultured with T cells and CRC antigen-pulsed dendritic cells (DCs) significantly abrogated T-cell responsiveness and induced the expansion of immunosuppressive regulatory T cells (Tregs). In addition, portal injections of HSCs with tumor cells significantly increased CRLM compared to tumor cells alone [44][26].
2.1.3. Angiogenic Phase
CRLMs are predominantly supplied by hepatic arteries [45][27]. The vascularization of liver metastases can occur through vascular endothelial growth factor (VEGF)-mediated angiogenesis or by the “co-opting” of existing vasculature. Importantly, a phase III trial by Hurwitz et al. [46][28] showed that the addition of bevacizumab, an anti-VEGF agent, to standard chemotherapy improved the median survival of patients with mCRC; however, the majority of patients did not exhibit a response to therapy [46][28]. This suggests the importance of other mechanisms of tumor vascularization independent of VEGF in some CRCs [42,47][24][29]. Notably, Frentzas et al. [48][30] described the co-opting of vessels in CRLM, a process by which cancer cells invade along the existing liver microvasculature, replacing hepatocytes and occupying the space adjacent to sinusoidal vessels. This commonly occurs in tumors that display a replacement growth pattern (see Section 3.1.4. below below) and is associated with resistance to anti-VEGF therapy. Importantly, the inhibition of actin-related protein 2/3 (Arp2/3), which is associated with the co-opting of vessels, was found to reduce vascular density in combination bevacizumab significantly more than bevacizumab by itself. Combination therapy also potently inhibited the growth of CRLMs with reduction in lesion area by approximately 75% compared to controls [48][30].
2.1.4. Growth Phase
The growth of vascularized micrometastases leads to tumor cell proliferation and the establishment of clinically detectable metastases. Three histologic growth patterns have been described by Eefsen et al. [25][7] in a series of 24 patients with resected CRLMs: desmoplastic, pushing, and replacement. In the desmoplastic pattern, a band of fibrotic tissue separates tumor cells from the surrounding liver parenchyma. In the pushing pattern, liver cells are pushed aside by the growing metastatic lesion and assume a flattened morphology with no fibrotic band separating the invasive tumor front from the hepatic parenchyma. The replacement pattern describes tumor cell infiltration into the existing liver parenchyma, such that it supplants the parenchyma instead of simply pushing it away. In 83% of patients, identical growth patterns were identified in all metastases [25][7]. This classification is notable because the dominant replacement growth pattern is associated with a worse prognosis [49][31]. These differences indicate that different interactions of tumor cells, TME cells, and cells of the hepatic parenchyma lead to varying CRLM phenotypes.
3. Colorectal Lung Metastasis (CRPM)
The lungs are the second most common site of CRC metastasis. Studies suggest that 10–18% of rectal cancers and 5–6% of colon cancers metastasize to the lungs [50][32]. According to population-based studies, the incidence of synchronous CRPMs has been estimated to be up to 11%, and has increased over time [50,51,52][32][33][34]. Similar to CRLM, specific oncogenes are implicated in CRPM. Zhang et al. [53][35] demonstrated through extensive genomic profiling studies of clinical samples from five CRC patients that CRPM shared clonality with primary tumors. Furthermore, 14 of 27 mutated genes identified in CRPM (i.e., KRAS, APC, and TP53) were also found in the primary lesion, thus suggesting that these mutations likely existed in the primary lesion itself. As such, the identification of these mutations in the primary tumor during diagnostic workup or on post-resection pathologic analysis may be prognostically significant [53][35]. While surgical resection for isolated CRPMs is the only curative option for select patients, there are no high-quality clinical-trial data to guide practice. For most patients, systemic therapy is the cornerstone of treatment. Consistent with this, in their decellularized liver and lung scaffolding models, Tian et al. [28][10] found that CRPM-model cells were significantly more sensitive to chemotherapy compared to CRLM-model cells, suggesting that unique microenvironment features affect therapeutic response. As such, an improved understanding of the TME of CRPM may identify new therapeutic targets.
Pathophysiology of Colorectal Metastasis to the Lungs
CRCs metastasize to the lungs through different mechanisms. Lymphatic vessels from the colon and proximal rectum drain into the inferior mesenteric nodes, which follows venous drainage into the portal circulation. Outflow from the distal rectum through the middle and inferior rectal veins drains into the internal iliac veins and directly into systemic circulation, thereby bypassing the portal system. Distal rectal lymphatics follow a similar course into the internal iliac nodes. As such, distal rectal tumors are more likely to present with early CRPM due to direct systemic drainage, as opposed to proximal tumors that must first filter through the liver [51][33]. Additionally, lymphatic vessels drain into the cisterna chyli and ultimately into the left subclavian vein through the thoracic duct, which may also lead to the development of CRPM [54][36].
The microenvironment of the lung parenchyma is enriched with a diverse array of immune cells that reside along the airway, including alveolar macrophages (dust cells), lymphocytes, and DCs, which are crucial for defense against airborne pathogens, toxins, and inflammatory substances [55,56][37][38]. Chronic inflammatory conditions, such as cigarette smoking and chronic obstructive pulmonary disease, alter the microenvironment in a way that lends itself to primary tumor development and the establishment of a pre-metastatic niche [57,58,59][39][40][41]. Indeed, current smoking status was found to be an independent risk factor for CRPM in a multivariable analysis of outcomes from 567 CRC patients [60][42].
Clearly, CRPMs (as well as other metastatic sites) represent an escape from immune surveillance. Thus, it is of great interest that a key feature of lung metastases is a tendency toward having a more immune responsive TME relative to metastases in other organs, such as the brain, liver, or bone [61][43]. García-Mulero et al. [61][43] analyzed resected tumor tissue and found that lung metastases from various primary tumors, including CRC, were enriched for genes associated with antigen presentation and immune effector cells, such as cytotoxic T lymphocytes (CTLs) and B-lineage cells. Moreover, they observed a low density of suppressor cells [61][43]. Lung metastases had a higher infiltration of CTLs, B-lineage cells, and DCs, but also expressed high levels of PD-L1 and cytotoxic T-lymphocyte–associated protein 4 (CTLA-4), which could partly explain their ability to escape immune surveillance [61][43]. However, infiltration by neutrophils, NK cells, and myeloid lineage cells was similar to other metastatic sites [61][43]. The overall high immunogenicity of the lung metastatic TME, as well as the elevated expression of PD-L1 and CTLA-4 in these lesions, may confer increased sensitivity to ICIs.
Altkori et al. [56][38] described a general mechanism for the formation of lung metastasis from extra-thoracic tumors. First, primary tumors secrete extracellular vesicles and pro-metastatic factors, including TGF-β, VEGF, and others that remodel the ECM, promote epithelial–mesenchymal transition (EMT), and facilitate invasion into the systemic circulation. These factors also promote recruitment of bone-marrow-derived cells into the microenvironment. Circulating tumor cells then extravasate into the pulmonary tissue. Type II alveolar cells recruit neutrophils, which suppress CTL activity and work with fibroblasts to facilitate seeding of tumor cells into the lung parenchyma. In addition, macrophages in the metastatic niche promote tumor cell survival and proliferation. Altogether, the established metastatic niche sustains tumor cell growth, promotes mesenchymal–epithelial transition, and stimulates angiogenesis [56][38].
4. The Tumor Microenvironment in CRC
To strengthen our understanding of the pathophysiology of CRLM and CRPM, it is crucial to first investigate in detail the function of individual components of the TME in CRC and their contribution to primary tumor invasiveness and subsequent metastasis to the liver and lung. The TME is the environment surrounding tumor cells and includes a complex array of immune cells, fibroblasts, endothelial cells, and stromal proteins. It is implicated in virtually every aspect of tumor progression and the metastatic cascade, including initial engraftment, growth, and immune evasion. Here, we review the function of selected cell types of the CRC TME (Figure 2).
4.1. Immune Cells
4.1.1. Macrophages
Tumor-associated macrophages (TAMs) have historically been dichotomized as “M1-like” macrophages with pro-inflammatory, immunostimulatory, and anti-tumorigenic properties or “M2-like” macrophages with immunosuppressive and pro-tumorigenic properties [62][44]. In reality, TAMs exist on a spectrum between these two states; however, this traditional classification provides a useful framework for discussion. M1 macrophages produce pro-inflammatory immunostimulatory cytokines, including IL-1β, IL-6, and TNF-α [62,63][44][45]. Although the M1 phenotype is generally thought to be anti-tumorigenic, this notion is context-specific. For example, M1 TAMs can promote chronic inflammation in induced colitis models, and this is a risk factor for the development of CRC [64][46]. In a colitis-associated CRC model, Wang et al. [65][47] demonstrated that the densities of M1 and M2 macrophages vary throughout the inflammation–carcinoma sequence, but overall macrophage count increased as tumors became metastatic. Exposure of CRC cell lines to conditioned media from M1 TAMs decreased viability and promoted pro-apoptotic morphologic changes, while treatment with M2-derived conditioned media increased cell proliferation and expression of the anti-apoptotic markers survivin and BMI-1 compared to control conditioned media [66][48]. Activated M2 TAMs also contribute to tumor angiogenesis through secretion of VEGF along with other TME cells [67,68,69][49][50][51].
M2 TAMs promote invasion and metastasis [70,71,72,73,74][52][53][54][55][56]. TAM secrete of MMP-9, a key proteolytic enzyme that contributes to ECM remodeling in multiple cancers, including CRC [72][54]. Concordantly, Afik et al. [74][56] demonstrated that CRC TAMs were enriched for molecular signatures associated with ECM remodeling in transcriptomic and proteomic analyses [74][56]. M2 TAMs also contribute to the production of TGF-β, which potentiates ECM remodeling, EMT, and metastasis [71][53]. EMT pertains to tumor cell loss of epithelial features (i.e., the loss of apical–basal polarity, dissolution of tight junctions, etc.) and assumption of a mesenchymal-like phenotype [75][57]. The mesenchymal phenotype is associated with increased invasiveness and migratory potential [75][57]. The loss of E-cadherin expression, a hallmark of the epithelial phenotype, is associated with a poor prognosis in CRC [76][58]. TAMs also promote EMT through IL-6-mediated activation of the JAK2/STAT3 pathway and subsequent inhibition of the miRNA tumor suppressor miR-506-3p and its target, FoxQ1, as well as through secretion of CCL22 and subsequent activation of the PI3K/AKT pathway [73,77][55][59].
M2 TAMs also facilitate immune evasion through secretion of immunosuppressive cytokines IL-10 and TGF-β, which facilitate suppression of T lymphocytes [65,78][47][60]. Herbeuval et al. [79][61] demonstrated that TAMs induced an immunosuppressive environment through the secretion of IL-6, leading to STAT3-mediated production of IL-10 by CRC cells. Thus, M2 TAMs in the CRC TME drive tumor progression, invasiveness, and a more aggressive tumor phenotype.
TAMs in CRLM and CRPM
TAMs are directly implicated in CRLM and may have context-specific pro-metastatic and anti-metastatic effects. In a microarray analysis of 159 resected CRLM specimens, the presence of CD68+ TAMs was associated with longer disease-free survival (DFS) on multivariable analysis [80][62]. Conversely, Wang et al. [81][63] demonstrated that CD206+ M2 TAMs were positively correlated with the presence of CRLM in patient samples; activation of the CXCL12/CXCR4 axis in CRC tumor cells promoted the exosome-mediated release of PTEN-suppressing miRNAs (miR-25-3p, miR-130b-3p, and miR-425-5p), which polarized TAMs to an M2 phenotype. Notably, murine tumors derived from HCT116 and MC38 CRC cells plus macrophages transfected with these miRNAs significantly increased tumor volume, and tail vein co-injection resulted in a significantly greater number and size of CRLM nodules compared to an injection of tumor cells alone [81][63]. KCs may also contribute to CRLM through the promotion of ECM remodeling and facilitation of tumor cell invasion for those CRC cells that escaped initial phagocytosis [43][25]. In this sense, they may serve a protective role against tumor cell infiltration earlier during metastasis. Indeed, Wen et al. demonstrated that the depletion of KCs at the earlier exponential growth phase increased CRLM burden in orthotopic murine models, while delayed KC depletion restricted tumor burden. This suggests that the pro- or anti-metastatic function of KCs may be time-dependent [82][64]. KCs and TAMs may also be impacted by CRLM resection. In a murine model of associated liver partition and portal vein ligation for staged hepatectomy for CRLM, mice who underwent this combined procedure had a significantly higher infiltration of KCs, which were polarized to the M1 phenotype, while TAMs were polarized to the M2 phenotype. This suggests that M1 KCs may be involved in a reactive inflammatory response, and that TAMs, perhaps M2-polarized as a consequence of induced hypoxia after the procedure, may contribute to persistent tumor progression and mixed therapeutic response [83][65].
How TAMs directly contribute to CRPM formation is not as well-elucidated. Cai et al. [71][53] demonstrated in CT26 CRC cell line–derived xenografts that the injection of TAMs into the tumor site significantly increased the number of CRPM nodules and total lung weight, a process dependent on TAM-derived TGF-β secretion and the induction of EMT through the modulation of the Smad2/3/4–Snail–E-cadherin pathway. Recently, KRAS mutational status was identified as a metastatic tropic factor for CRPM development, significantly more so than CRLM [84][66]. Patients with mutant KRAS tumors had a significantly shorter time to CRPM development and CRPM-free survival compared to those wild-type KRAS status [84][66]. Concordantly, Liu et al. [85][67] demonstrated that mutant KRAS reprograms macrophages to a M2 TAM-like phenotype through colony-stimulating factor 2 and lactate production, which promoted tumor progression and conferred EGFR inhibitor resistance.
4.1.2. Helper T Cells
CD4+ helper T cells are key mediators of the adaptive immune system, secreting cytokines to modulate the activity of other immune cells in response to infection or cancer. The presence of tumor-infiltrating lymphocytes (TILs) is associated with improved survival in CRC. In a study of 342 resected CRC specimens, high CD4+ T-cell density was associated with improved relapse-free survival and disease-specific survival [86][68]. However, CD4+ T-cell subtypes in the TME have varying impacts on tumor behavior. T-helper 1 cells (TH1) are traditionally thought to enhance CTL effector function and are instrumental for augmentation of the antitumor immune response [87][69]. Increased expression of TH1-cluster genes in resected CRC specimens is associated with improved disease-free survival, while higher numbers of T-helper 17 cells (TH17), which have an immunosuppressive function, are associated with poor survival [88][70]. This is consistent with prior preclinical studies, which suggest that TH17-derived cytokines, such as IL-17, IL-21, and IL-22, promote CRC tumor growth [89][71].
Helper T Cells in CRLM and CRPM
Kroemer et al. [90][72] demonstrated that the presence of expanded TH17 CD4+ T cells in resected CRLM was associated with poor prognosis. At the tumor site, while CD4+ T cells may be the most frequently encountered T-cell population, their ability to proliferate in response to tumor antigens after DC-mediated pulsing was significantly diminished [91][73]. These findings are consistent with those from Katz et al. [92][74], who demonstrated that high CD4+ T-cell density was inversely associated with survival after CRLM resection. In another study, Katz et al. [93][75] demonstrated through murine models that hepatic CD4+ T cells are generally polarized toward the TH2 phenotype and produced immunosuppressive cytokines. This may, in part, explain why the presence of CD4+ T cells negatively impacts prognosis. Little is known about how CD4+ T cells specifically impact CRPM. In other lung metastasis models including breast cancer, TH2-CD4+ T cells have been shown to augment metastasis through the potentiation of macrophage activity [94][76]. Further investigation into how CD4+ T cells specifically impact CRPM is necessary.
4.1.3. Dendritic Cells (DCs)
DCs are professional antigen-presenting cells that are crucial for the initiation of the immune response to specific antigens through internalization of foreign antigens and subsequent presentation to T cells. To avoid immune surveillance, cancer cells may suppress DCs through multiple mechanisms, including the secretion of immunosuppressive TGF-β [95][77]. In CRC, myeloid DCs, the most common DC subtype associated with cell-mediated immunity and stimulation of naïve CD4+ T cells to the TH1 phenotype, are found in increased frequency at the tumor invasive front and are associated with lymph node invasion [96][78]. In contrast, Gulubova et al. [97][79] noted that advanced-stage CRC tumors had a lower density of CD83+ DCs in the stroma and invasive margin [97][79].
The mechanism through which these DCs enhance invasiveness has not been well-elucidated. Orsini et al. [98][80] demonstrated that DCs from CRC patients had impaired antigen-presenting capacity and reduced co-stimulatory molecule expression compared to DCs from healthy controls. Furthermore, CRC patient-derived DCs secreted increased levels of immunosuppressive IL-10 and decreased levels of immunostimulatory IL-12 and TNF-α [98][80]. Concordantly, Nagorsen et al. [99][81] demonstrated that while S100+ DCs were found more frequently in limited disease and were associated with improved survival, their presence in the CRC TME was also positively associated with Treg infiltration, further highlighting the paradoxical pro-tumorigenic function of CRC-hijacked DCs. Indeed, Hsu et al. [100][82] demonstrated that DCs harvested from CRC patients highly expressed CXCL1, which enhanced tumor cell migration, cancer cell stemness, and EMT. Thus, DC immunostimulatory capacity may be suppressed by CRC cells and modulated into an invasive, immunosuppressive phenotype, perhaps reflecting a reversion back to an immature phenotype [101][83].
DCs in CRLM and CRPM
Enhancing the DC presence in the CRLM TME may be a therapeutic target. Fused allogenic peripheral blood DCs with SW620 CRC cells promoted significantly higher secretion of IFN-γ by CD8+ T cells and, when injected into highly immunodeficient CRLM mouse models, markedly reduced tumor growth compared to sham-vaccinated CRC cells [102][84]. These findings are consistent with prognostic data suggesting that the absence of CD83+ DCs in resected CRLM specimens is associated with worse 5-year OS, partially due to diminished antigen presentation and immunostimulation [103][85]. This vaccination strategy was being investigated in a CRLM clinical trial which, despite being terminated early, demonstrated evidence of improved median DFS [104][86].
Hsu et al. [100][82] demonstrated that CXCL1 secretion by DCs augments CRPM. The sequencing of CXCL1-enriched SW620 CRC cells resulted in enrichment of prognostically significant genes including PTHLH (parathyroid hormone-like hormone). When these cells were implanted into murine livers, they were found to have a heightened capacity for CRPM formation compared to native SW620 cells, which was dependent on suppression of the p38/MAPK pathway and subsequent upregulation of PTHLH [105][87]. The inhibition of PTHLH selectively inhibited CRPM formation from CRLM, but not the growth of directly implanted cells or of liver metastases themselves [105][87].
4.1.4. Regulatory T Cells (Tregs)
Tregs, characterized by CD25 and FoxP3 expression, are potent mediators of immunosuppression. Their presence in the TME is associated with increased metastasis and poor outcomes in numerous malignancies [106,107,108][88][89][90]. In their physiologic state, Tregs prevent autoimmunity and regulate the immune response by downregulating IL-2, releasing adenosine, and secreting immunosuppressive cytokines including TGF-β, IL-10, and IL-35 [109][91]. In CRC, the impact of Tregs in the TME is complex, and evidence suggests their function as pro- or anti-tumorigenic is context-specific. Ji et al. [110][92] demonstrated through mRNA profiling of 81 pretreatment biopsies of locally advanced rectal cancer patients that the Tregs were inversely correlated with prognosis. Conversely, other studies demonstrate that elevated FoxP3+ cell infiltration correlate with improved survival [86,111][68][93].
A potential explanation for the apparent contradictory effect of Tregs is likely due to their heterogeneity. Saito et al. [112][94] demonstrated that naïve Tregs (FoxP3low/CD45RA+) in CRC specimens could be stratified into terminally differentiated immunosuppressive FoxP3high/CD45RA-, and pro-inflammatory FoxP3low/CD45RA- subgroups. Compared to other tumors, such as melanoma, some CRC specimens had significantly higher numbers of FoxP3low/CD45RA- Tregs [112][94]. The authors therefore characterized CRC tumors as “type A” with a low expression of these inflammatory Tregs or “type B” with a high expression of inflammatory Tregs. Notably, type-B CRC demonstrated significant upregulation of genes involved with inflammation and immune response, including IL-12, TNF-β, and TGF-β [112][94]. Consistent with these findings, high FOXP3 expression in type-A CRC tumors was associated with a worse prognosis [112][94].
Regulatory T Cells in CRLM and CRPM
While Treg heterogeneity has not been investigated in detail specifically for CRLM, their overall presence markedly impacts prognosis. Pedroza-Gonzalez et al. [91][73] demonstrated that CRLMs contain sequestered activated Tregs at the tumor site, which highly expressed glucocorticoid-induced TNF receptor (GITR) and were more potently immunosuppressive compared to Tregs from primary hepatocellular carcinoma. The ablation of GITR prevented Treg-mediated suppression of effector T-cell activity, suggesting a possible immunostimulatory therapeutic target [91][73]. Katz et al. demonstrated immunohistochemical analysis of 188 resected CRLM specimens that higher ratios of FoxP3+ Treg:CD4+ and FoxP3+ Treg:CD8+ ratios were associated with shorter 5-year overall survival compared to lower ratios (34% vs. 51%, and 35% vs. 46%, respectively) [113][95].
Tregs are prevalent in CRPM, but little is known about specific functions in this niche [114][96]. Resident Tregs may contribute to the pulmonary pre-metastatic niche. In physiologic states, pulmonary tissue is enriched with Tregs, which are crucial in regulating immune responses to inhaled environmental antigens. Through many of the previously discussed mechanisms, pulmonary Tregs may promote immunosuppression in the lung microenvironment and foster a niche for pulmonary metastasis [115][97].
4.1.5. Neutrophils
Neutrophils are the first responders to acute infection or inflammation [116][98]. They secrete lytic enzymes, phagocytose certain microbes, generate neutrophil extracellular traps, and secrete numerous cytokines that sustain the inflammatory response and recruit other immune cells as part of the innate immune response. Tumor-associated neutrophils (TANs) are associated with prognosis; as such, understanding the function of TANs is an emerging area of investigation [117][99]. Fridlender et al. [118][100] demonstrated that TGF-β production in the CRC TME polarizes TANs from an immunostimulatory “N1” phenotype to a pro-tumorigenic “N2” phenotype. TANs promote invasiveness through modulation of angiogenesis and resistance to VEGF inhibition. In a colitis-associated CRC mouse model, Itatani et al. [119][101] demonstrated that the induction of colitis increased granulocyte colony stimulating factor (G-CSF) levels in anti-VEGF therapy-resistant tumors, which was associated with increased Bv8/PROK2+ neutrophil infiltration. The dual inhibition of G-CSF or its receptor along with VEGF inhibition resulted in significantly suppressed tumor formation and angiogenesis [119][101].
TANs also contribute to immune evasion. In an inducible CRC murine model, Germann et al. [120][102] demonstrated that presence of T cells in adenomas was diminished and inversely correlated with neutrophil infiltration. These TANs were enriched for immunosuppressive gene expression and secreted MMP-9, which suppressed T-cell proliferation through the activation of TGF-β [120][102]. Overall, TANs may also facilitate the dissemination of tumor cells by augmenting degradation of the basement membrane, which enhances tumor mobility and promotes tumor cell extravasation [121][103].
TANs in CRLM and CRPM
TANs contribute to the growth of CRLM through multiple mechanisms. In murine models, Gordon-Weeks et al. [122][104] demonstrated that CRLM-associated neutrophils were enriched in tumors, expressed high levels of fibroblast growth factor-2 (FGF2), and enhanced the growth of CRLM in mice. The depletion of neutrophils 7 days after an intrasplenic injection of CRC cells diminished CRLM development and angiogenesis. Monoclonal antibody (GAL-F2) blockade of FGF2 also restricted tumor growth and normalized vasculature, suggesting an FGF2-dependent mechanism through which TANs contribute to CRLM [122][104]. This is consistent with the previously discussed findings of Itatani et al., who also demonstrated that granulocyte-colony stimulating factor (G-CSF) secretion by CRC cells enhanced TAN recruitment in CRLM as well as in primary CRC tumors, and conferred similar resistance to anti-VEGF therapy [119][101]. Additionally, Yang et al. [123][105] demonstrated that neutrophils derived from CRC patient sera prominently secreted neutrophil extracellular traps (web-like consolidations of chromatin and proteases thought to be released by activated neutrophils and believed to enhance tumorigenesis perhaps through trapping cells at metastatic sites), which correlated with the risk of CRLM. Furthermore, CXCL8 secretion from CRC cells mediated the production of neutrophil extracellular traps, which enhanced CRLM formation in vivo [123][105].
A combination of SMAD4 deficiency in CRC cells plus CCR1+ TANs is implicated in the progression of CRPM [124][106]. SMAD4 is a tumor suppressor, and its inactivation is associated with multiple cancers, including CRC [125,126,127][107][108][109]. SMAD4-deficient CRC cells secrete higher levels of CCL15, which recruits CCR1+ TANs through the CCL15-CCR1 chemokine axis to promote CRPM [124][106]. TANs are also critical drivers for the development of the pre-metastatic niche at secondary sites like the lung [128,129,130][110][111][112]. In a model of melanoma pulmonary metastasis, El Rayes et al. [59][41] demonstrated that LPS-inflamed lungs were enriched for Ly6G+ neutrophils and developed a greater metastatic burden compared to controls. These TANs proteolytically degraded thrombospondin-1 (Tsp-1), an ECM protein that regulates inflammation, inhibits angiogenesis, and restricts tumor growth [59][41].
4.1.6. Cytotoxic T Cells
CTLs are the primary effector cells of the adaptive immune response and are a crucial component of antitumor immunity. The presence of CTLs in patient-derived CRC tumor cores is associated with a reduced risk of recurrence, particularly in higher-stage tumors [131][113]. Concordantly, the CRC Immunoscore prognostic tool was developed to supplement the TNM staging paradigm and is based on the prevalence of infiltrating CD3+ cells and CTLs. Higher Immunoscore values positively correlate with improved survival and are corroborated by internal and external validation studies [132,133][114][115].
To promote immune evasion, CTL activity is suppressed through multiple mechanisms. O’Malley et al. [134][116] demonstrated that TNF-α–mediated inflammation in CT26 CRC cells induced PD-L1 expression in stromal cells, resulting in the inhibition of activated granzyme-secreting CD8+ T cells [134][116]. Tumor and stromal cells express PD-L1, which, upon binding to the PD-1 receptor on T cells, suppresses T-cell activation. In CRC, a higher expression of PD-L1 is associated with higher stage, higher grade, lymph node involvement, distant metastasis, and reduced overall survival [135,136,137][117][118][119]. The PD-1/PD-L1 axis is an attractive target to increase immune targeting. Indeed, PD-1 blockade with pembrolizumab was FDA-approved for MSI-high/mismatch repair-deficient subtype advanced tumors in the pivotal KEYNOTE 177 trial [4,138][4][120]. Other TME cells also curtail CTL function through TGF-β secretion, which inhibits CTL expression of the key cytolytic enzymes: perforin, granzyme, and Fas ligand [139][121]. Furthermore, Tregs recruited via the CCL5/CCR5 signaling pathway have a heightened ability to kill CTLs in CRC murine models [140][122].
CTLs in CRLM and CRPM
CRLMs are more highly enriched for PD-L1 expression compared to primary tumors, particularly rectal cancer liver metastases [29][11]. In a study of 74 patients with resected CRLM and primary tumors, Wei et al. [29][11] found that high PD-L1 expression was significantly associated with an enrichment of CD4+ T cells and CTLs. Concordantly, Katz et al. [92][74] observed that a high density of CTLs positively correlated with 10-year survival after CRLM resection, and on multivariable analysis, a high CD8+/low CD4+ phenotype was significantly associated with better long-term survival.
Similarly, Remark et al. [141][123] found that an increased presence of LAMP+ DCs and CTLs was positively associated with improved overall survival in CRPM, but not in renal cell carcinoma lung metastases, suggesting a context-specific function. Furthermore, the density of CTLs, as well as DCs and NKs, in primary CRC tumors correlated with densities in CRPM, suggesting a reproducible immune pattern from the primary tumor possibly secondary to TME “imprinting” from the primary site or the development of educated immune cells [141][123]. The authors suggest that, since tertiary lymphoid structures are prevalent in CRPM, these T cells may be more educated compared to renal cell carcinoma metastases, which tend to have a scarce presence of tertiary lymphoid structures [141][123]. This may point toward a sensitivity of CRPM and CRLM to ICIs, particularly in patients with MSI-high/mismatch repair-deficient subtype advanced CRC [4,138][4][120].
4.2. Stromal Cells and the Extracellular Matrix
4.2.1. Extracellular Matrix
The ECM comprises structural proteins, including collagen, proteoglycans, hyaluronic acid, and glycosaminoglycans, whose structure and function can be remodeled by cells in the TME [142][124]. In addition to being the physical scaffolding for tumor cells, the ECM dynamically contributes to cell–cell adhesion, paracrine signaling, tumor proliferation, immune evasion, and metastasis [143][125]. Remodeling of the ECM is a consistent feature of the TME in many indications including CRC. Yang et al. [144][126] demonstrated that density of the remodeling enzyme MMP-9 was markedly greater in CRC compared to normal mucosa and higher expression was associated with worse prognosis. MMP-2 has also been implicated in ECM degradation and correlates with lymphatic invasion and advanced stage [145,146][127][128]. MMP-2 and MMP-9 encode collagenases that target type IV collagen, which is found in the basement membrane; and the degradation of type IV collagen can enhance tumor cell motility and invasion [145,147,148][127][129][130]. The biomechanics of the ECM also contribute to CRC proliferation. Using atomic force microscopy, Brauchle et al. [149][131] demonstrated that collagen-rich regions of the CRC ECM were stiffer compared to normal tissue. Stiffness of CRC has been associated with metastasis and EMT [150][132]. Li et al. [143][125] observed that the density of type I collagen positively correlated with stage, while type IV collagen density was reduced in higher stage tumors. This latter finding is consistent with invasion through the basement membrane [151][133]. The inverse relationship between density of type IV collagen and CRC stage likely reflects increased ECM remodeling, partly through MMP-2 and -9 activity [147][129]. At distant sites, proteomic analyses of control vs. matched CRLM patient tissues suggest that, while the ECM of CRLM more closely resembles the primary tumor compared to liver parenchyma, it is enriched for several unique proteins (i.e., SPP1 and COMP) [152][134]. Yuzhalin et al. [153][135] found that the CRLM ECM had higher levels of citrullinating enzyme peptidylarginine deiminase 4 (PAD4) compared to benign liver tissue, primary CRC, or colonic mucosa. CRC cells grown in citrullinated type I collagen exhibited mesenchymal-to-epithelial transition (reversion to a proliferative epithelial phenotype of tumor cells) in vitro and enhancement of CRLM growth in vivo [153][135].
4.2.2. Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs) contribute significantly to ECM maintenance, desmoplasia, angiogenesis, immunosuppression, invasion, and chemoresistance [154,155][136][137]. While a majority of CAFs arise from resident stromal fibroblasts, mesenchymal stem cells (MSCs) and mesothelial cells also contribute a significant proportion of the CAF progenitor population [154,156][136][138]. Bone-marrow-derived MSCs, which are pluripotent stem cells involved in tissue remodeling, may be recruited to tumor sites to aid in tumor growth and progression [157][139]. Once localized at the tumor site, MSCs are induced by tumor-derived factors to assume a CAF-like phenotype [158][140]. Meanwhile, mesothelial cells contribute to the CAF population in highly invasive tumors via mesothelial-to-mesenchymal transition, which has been implicated in CRC peritoneal carcinomatosis [159][141]. Overall, in CRC, two major subpopulations of CAFs have been identified through single-cell RNA sequencing: CAF-A (which express ECM remodeling-associated genes, including MMP2, DCN, and COL1A2) and CAF-B (myofibroblast-like that express ACTA2, TAGLN, and PDGFA) [160][142].
CAFs promote immune evasion [120,154,161,162][102][136][143][144]. In microsatellite stable CRC samples, Tauriello et al. demonstrated an inverse correlation between the immunostimulatory TH1 to naïve T-cell ratio and the mean expression of CAF-specific genes [162][144]. Tumor invasive margins had high levels of stromal TGF-β, of which CAFs were the primary source. The elevated expression of TGF-β in the TME inhibited TH1 T-cell function. Additionally, CAFs secrete CXCL8, which attracts monocytes to the CRC TME and promotes M2 polarization, further contributing to immunosuppression [163][145].
CAFs promote metastasis through ECM remodeling and EMT in multiple cancers, including CRC [164,165,166,167][146][147][148][149]. Their presence in the CRC intratumoral stroma is associated with lymphatic invasion [168][150]. Through the secretion of ECM remodeling enzymes, collagen, and other cytoskeletal proteins, CAFs promote desmoplasia, which can be identified in up to 78% of CRC tumor specimens [168][150]. TGF-β/Smad2 signaling is prominently activated in CAFs, and CRC cells themselves can stimulate TGF-β production in CAFs with resulting expression of α-SMA and differentiation to a myofibroblastic phenotype [169][151]. These upregulate the expression of invasion-related proteinases, including multiple MMPs [169][151]. Notably, TAMs may regulate CAF activity in the CRC ECM with respect to the expression of collagen types I and XIV [74][56]. The expression of collagen crosslinking enzyme lysyl oxidase–like 2 (LOXL2) by CAFs in CRC tumors is associated with a higher rate of recurrence and worse overall survival and disease-free survival [170][152]. Through LOXL2, CAFs also stimulate EMT through activation of the FAK pathway, with a resultant reduction in E-cadherin protein expression. CRC cells co-cultured with these activated fibroblasts demonstrated increased migration rates [167][149]. CAFs also promote angiogenesis through secretion of IL-6; Nagasaki et al. [171][153] showed that CRC cells potentiated IL-6 secretion from CAFs with resulting upregulation of VEGFA expression. Thus, CAFs contribute to CRC progression through immunosuppression, ECM remodeling, and the promotion of EMT.
CAFs in CRLM and CRPM
In murine models, CRLM tumor cells recruit CAFs to the metastatic site, which contributes to tumor progression [172][154]. Circulating levels of TGF-β are a predictor of future development of CRLM after resection of the primary tumor [173][155]. Calon et al. [174][156] demonstrated that increased stromal TGF-β signaling significantly promoted CRPM and CRLM in murine models through the upregulation of GP130–p-STAT3 signaling in CRC cells. TGF-β–stimulated CAFs in the CRC TME secreted IL-11, a key ligand for GP130, resulting in the upregulation of anti-apoptotic factors MCL-1 and Bcl-2 [174][156]. Intrasplenic injection of HT29-M6 cells autonomously producing IL-11 more robustly colonized the liver and exhibited reduced apoptosis within the first hours of colonization compared to controls [174][156]. CAFs also potentiate metastasis through secretion of hepatocyte growth factor (HGF). Zhang et al. [175][157] demonstrated that secretion of HGF by CAFs upregulated CD44, and enhanced adhesive and migratory capacity of CRC cells in vitro and increased CRLM and CRPM formation in vivo. Resident fibroblasts also contribute to CRLM formation as well as the TME CAF population. As discussed, activated HSCs secrete ECM components and provide scaffolding for newly seeded CRC cells and contribute to immune evasion [44][26]. Furthermore, HSC-derived myofibroblasts secrete stromal-cell-derived factor 1 (a CXCR4 ligand), which can promote primary tumorigenesis in nude mice. Concordantly, CXCR4 may be a therapeutic target, as its inhibition with AMD3100 significantly diminished CRLM [176][158]. HSCs may not be the primary progenitors for CRLM CAFs, but rather resident portal fibroblasts. Mueller et al. [177][159] demonstrated that CRLM CAFs are similar to resident portal fibroblasts in their myofibroblastic phenotypes (α-SMA+, ICAM-1+, Thy-1+) and do not closely resemble HSCs (in contrast to the HSC phenotype, they are negative for glial fibrillary acidic protein, desmin, and neural cell adhesion molecules).
CAFs impact the prognosis for CRPM. In a study of 181 CRC-patient primary-tumor and metastatic-site specimens, Kwak et al. [178][160] demonstrated that CRPM had lower density of CAFs compared to primary tumors, but similar to the density at other tumor sites. The presence of CAFs at these metastatic sites and the loss of their PTEN expression were significantly associated with a worse prognosis [178][160]. CAFs in the CRPM TME prominently secrete heat-shock protein-27 (Hsp27), a protein implicated in angiogenesis, EMT, and fibroblast motility [179][161]. Hsp27 expression was associated with reduced disease-free survival and overall survival after CRPM metastasectomy [179][161].
4.2.3. Endothelial Cells
Tumor vasculature prominently impacts CRC invasion and is an established therapeutic target [180,181][162][163]. Endothelial cells (ECs), along with pericytes, smooth muscle cells, and progenitor cells, comprise the vasculature [182][164]. While EC generally remain quiescent in healthy tissue, malignancy can induce vascular turnover and cell proliferation [183][165]. Local injury, hypoxia, and rapid malignant proliferation initiate paracrine signaling networks where factors such as VEGF, PDGF, CXCL8, and TNF-α secreted by multiple TME cells drive neovascularization to perfuse proliferating tumor tissue [184][166]. ECs are implicated in invasion and metastasis. Zhang et al. [185][167] demonstrated that expression of vascular cell-adhesion molecular 1 (VCAM1) expression in CRC was associated with more invasive features and poorer prognosis. Forced expression of VCAM1 in CRC cells promoted pseudopodia formation and increased trans-endothelial migration in vitro. Furthermore, after CRC cells have entered the systemic vasculature, circulating tumor-derived ECs have been recently identified that may reflect a unique EC state that contributes directly to tumor progression and metastasis [186][168].
Endothelial Cells in CRLM and CRPM
As discussed, LSECs are central in early metastatic seeding, which is driven by E-selectin mediated adhesion to the endothelium and VEGF-mediated lymphangiogenesis [37,187][19][169]. LSEC lectin (LSECtin) promotes migration in malignant colorectal cell lines in vitro and in vivo and inhibition of LSECtin reduced CRLM formation [188][170]. Additionally, tumor-activated LSECs secrete ICAM-1, which drives IL-1 production and decreased antitumor immune activity through impaired IFN-γ production and mannose receptor-dependent endocytosis [189][171]. LSECs have also been shown to induce chemotaxis and outgrowth in CRC by secreting macrophage inhibitory factors, whose levels correlate with CRLM development [190][172].
In CRPM, CUB domain-containing protein 1 (CDCP1) is a cell-surface marker which identifies colorectal circulating stem cells that exhibit tropism to the lung [191][173]. CDCP1 expression is associated with increased risk of CRPM formation and inferior metastasis-free survival, and it may be implicated in the seeding and adhesion of CTCs to the pulmonary vasculature [191][173]. Elevated VEGF expression in CRPMs has been identified as a prognostic marker which predicts lack of benefit from CRPM resection [192][174].
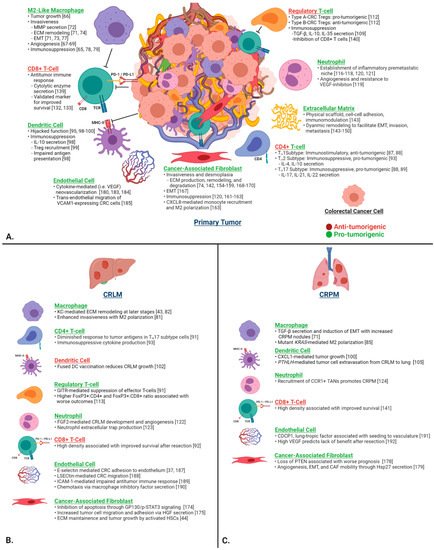
Figure 2. (A) Contributions of the key individual components of the CRC TME on tumor progression, invasion, ECM remodeling, immunosuppression, and metastasis in the primary tumor. (B) CRLM- and (C) CRPM-specific functions are depicted in the lower two panels. Green headings signify pro-tumorigenic functions, and red headings signify anti-tumorigenic functions. Abbreviations: CRC, colorectal cancer; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; MMP, matrix metalloproteinase; TME, tumor microenvironment.
References
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732.
- Kinj, R.; Bondiau, P.Y.; Francois, E.; Gerard, J.P.; Naghavi, A.O.; Leysalle, A.; Chamorey, E.; Evesque, L.; Padovani, B.; Ianessi, A.; et al. Radiosensitivity of Colon and Rectal Lung Oligometastasis Treated with Stereotactic Ablative Radiotherapy. Clin. Color. Cancer 2017, 16, e211–e220.
- Widder, J.; Klinkenberg, T.J.; Ubbels, J.F.; Wiegman, E.M.; Groen, H.J.; Langendijk, J.A. Pulmonary oligometastases: Metastasectomy or stereotactic ablative radiotherapy? Radiother. Oncol. 2013, 107, 409–413.
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218.
- Robinson, J.R.; Newcomb, P.A.; Hardikar, S.; Cohen, S.A.; Phipps, A.I. Stage IV colorectal cancer primary site and patterns of distant metastasis. Cancer Epidemiol. 2017, 48, 92–95.
- Horn, S.R.; Stoltzfus, K.C.; Lehrer, E.J.; Dawson, L.A.; Tchelebi, L.; Gusani, N.J.; Sharma, N.K.; Chen, H.; Trifiletti, D.M.; Zaorsky, N.G. Epidemiology of liver metastases. Cancer Epidemiol. 2020, 67, 101760.
- Eefsen, R.L.; Van Den Eynden, G.G.; Høyer-Hansen, G.; Brodt, P.; Laerum, O.D.; Vermeulen, P.B.; Christensen, I.J.; Wettergren, A.; Federspiel, B.; Willemoe, G.L.; et al. Histopathological Growth Pattern, Proteolysis and Angiogenesis in Chemonaive Patients Resected for Multiple Colorectal Liver Metastases. J. Oncol. 2012, 2012, 907971.
- Chu, P.C.; Lin, P.C.; Wu, H.Y.; Lin, K.T.; Wu, C.; Bekaii-Saab, T.; Lin, Y.J.; Lee, C.T.; Lee, J.C.; Chen, C.S. Mutant KRAS promotes liver metastasis of colorectal cancer, in part, by upregulating the MEK-Sp1-DNMT1-miR-137-YB-1-IGF-IR signaling pathway. Oncogene 2018, 37, 3440–3455.
- Brudvik, K.W.; Kopetz, S.E.; Li, L.; Conrad, C.; Aloia, T.A.; Vauthey, J.N. Meta-analysis of KRAS mutations and survival after resection of colorectal liver metastases. Br. J. Surg. 2015, 102, 1175–1183.
- Tian, X.; Werner, M.E.; Roche, K.C.; Hanson, A.D.; Foote, H.P.; Yu, S.K.; Warner, S.B.; Copp, J.A.; Lara, H.; Wauthier, E.L.; et al. Organ-specific metastases obtained by culturing colorectal cancer cells on tissue-specific decellularized scaffolds. Nat. Biomed. Eng. 2018, 2, 443–452.
- Wei, X.-L.; Luo, X.; Sheng, H.; Wang, Y.; Chen, D.-L.; Li, J.-N.; Wang, F.-H.; Xu, R.-H. PD-L1 expression in liver metastasis: Its clinical significance and discordance with primary tumor in colorectal cancer. J. Transl. Med. 2020, 18, 475.
- Giakoustidis, A.; Mudan, S.; Hagemann, T. Tumour microenvironment: Overview with an emphasis on the colorectal liver metastasis pathway. Cancer Microenviron. 2015, 8, 177–186.
- Brodt, P. Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin. Cancer Res. 2016, 22, 5971–5982.
- Milette, S.; Sicklick, J.K.; Lowy, A.M.; Brodt, P. Molecular Pathways: Targeting the Microenvironment of Liver Metastases. Clin. Cancer Res. 2017, 23, 6390–6399.
- Timmers, M.; Vekemans, K.; Vermijlen, D.; Asosingh, K.; Kuppen, P.; Bouwens, L.; Wisse, E.; Braet, F. Interactions between rat colon carcinoma cells and Kupffer cells during the onset of hepatic metastasis. Int. J. Cancer 2004, 112, 793–802.
- Matsumura, H.; Kondo, T.; Ogawa, K.; Tamura, T.; Fukunaga, K.; Murata, S.; Ohkohchi, N. Kupffer cells decrease metastasis of colon cancer cells to the liver in the early stage. Int. J. Oncol. 2014, 45, 2303–2310.
- Haier, J.; Korb, T.; Hotz, B.; Spiegel, H.-U.; Senninger, N. An intravital model to monitor steps of metastatic tumor cell adhesion within the hepatic microcirculation. J. Gastrointest. Surg. 2003, 7, 507–515.
- Gassmann, P.; Hemping-Bovenkerk, A.; Mees, S.T.; Haier, J. Metastatic tumor cell arrest in the liver–lumen occlusion and specific adhesion are not exclusive. Int. J. Color. Dis. 2009, 24, 851–858.
- Brodt, P.; Fallavollita, L.; Bresalier, R.S.; Meterissian, S.; Norton, C.R.; Wolitzky, B.A. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int. J. Cancer 1997, 71, 612–619.
- Khatib, A.-M.; Fallavollita, L.; Wancewicz, E.V.; Monia, B.P.; Brodt, P. Inhibition of hepatic endothelial E-selectin expression by C-raf antisense oligonucleotides blocks colorectal carcinoma liver metastasis. Cancer Res. 2002, 62, 5393–5398.
- Khatib, A.-M.; Auguste, P.; Fallavollita, L.; Wang, N.; Samani, A.; Kontogiannea, M.; Meterissian, S.; Brodt, P. Characterization of the host proinflammatory response to tumor cells during the initial stages of liver metastasis. Am. J. Pathol. 2005, 167, 749–759.
- Khatib, A.-M.; Kontogiannea, M.; Fallavollita, L.; Jamison, B.; Meterissian, S.; Brodt, P. Rapid induction of cytokine and E-selectin expression in the liver in response to metastatic tumor cells. Cancer Res. 1999, 59, 1356–1361.
- Lee, Y.; Friedman, S.L. Fibrosis in the Liver; Elsevier: Amsterdam, The Netherlands, 2010; pp. 151–200.
- Döme, B.; Hendrix, M.J.C.; Paku, S.; Tóvári, J.; Tímár, J. Alternative Vascularization Mechanisms in Cancer. Am. J. Pathol. 2007, 170, 1–15.
- Tsilimigras, D.I.; Brodt, P.; Clavien, P.-A.; Muschel, R.J.; D’Angelica, M.I.; Endo, I.; Parks, R.W.; Doyle, M.; De Santibañes, E.; Pawlik, T.M. Liver metastases. Nat. Rev. Dis. Primers 2021, 7, 1–23.
- Huang, W.-H.; Zhou, M.-W.; Zhu, Y.-F.; Xiang, J.-B.; Li, Z.-Y.; Wang, Z.-H.; Zhou, Y.-M.; Yang, Y.; Chen, Z.-Y.; Gu, X.-D. The role of hepatic stellate cells in promoting liver metastasis of colorectal carcinoma. OncoTargets Ther. 2019, 12, 7573.
- Breedis, C.; Young, G. The blood supply of neoplasms in the liver. Am. J. Pathol. 1954, 30, 969.
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342.
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436.
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.R.; Wotherspoon, A.; Gao, Z.-H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302.
- Fernández Moro, C.; Bozóky, B.; Gerling, M. Growth patterns of colorectal cancer liver metastases and their impact on prognosis: A systematic review. BMJ Open Gastroenterol. 2018, 5, e000217.
- Parnaby, C.N.; Bailey, W.; Balasingam, A.; Beckert, L.; Eglinton, T.; Fife, J.; Frizelle, F.A.; Jeffery, M.; Watson, A.J. Pulmonary staging in colorectal cancer: A review. Color. Dis. 2012, 14, 660–670.
- Mitry, E.; Guiu, B.; Cosconea, S.; Jooste, V.; Faivre, J.; Bouvier, A.M. Epidemiology, management and prognosis of colorectal cancer with lung metastases: A 30-year population-based study. Gut 2010, 59, 1383–1388.
- Van der Geest, L.G.; Lam-Boer, J.; Koopman, M.; Verhoef, C.; Elferink, M.A.; de Wilt, J.H. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin. Exp. Metastasis 2015, 32, 457–465.
- Zhang, N.; Di, J.; Wang, Z.; Gao, P.; Jiang, B.; Su, X. Genomic profiling of colorectal cancer with isolated lung metastasis. Cancer Cell Int. 2020, 20, 281.
- Riihimäki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6, 29765.
- Agostini, C.; Chilosi, M.; Zambello, R.; Trentin, L.; Semenzato, G. Pulmonary immune cells in health and disease: Lymphocytes. Eur. Respir J. 1993, 6, 1378–1401.
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31.
- Houghton, A.M. Mechanistic links between COPD and lung cancer. Nat. Rev. Cancer 2013, 13, 233–245.
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat. Rev. 2014, 40, 558–566.
- El Rayes, T.; Catena, R.; Lee, S.; Stawowczyk, M.; Joshi, N.; Fischbach, C.; Powell, C.A.; Dannenberg, A.J.; Altorki, N.K.; Gao, D. Lung inflammation promotes metastasis through neutrophil protease-mediated degradation of Tsp-1. Proc. Natl. Acad. Sci. USA 2015, 112, 16000–16005.
- Yahagi, M.; Tsuruta, M.; Hasegawa, H.; Okabayashi, K.; Toyoda, N.; Iwama, N.; Morita, S.; Kitagawa, Y. Smoking is a risk factor for pulmonary metastasis in colorectal cancer. Color. Dis. 2017, 19, O322–O328.
- García-Mulero, S.; Alonso, M.H.; Pardo, J.; Santos, C.; Sanjuan, X.; Salazar, R.; Moreno, V.; Piulats, J.M.; Sanz-Pamplona, R. Lung metastases share common immune features regardless of primary tumor origin. J. Immunother. Cancer 2020, 8, 1–12.
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in M1 (LPS+) vs. classically and M2 (LPS–) vs. alternatively activated macrophages. Front. Immunol. 2019, 10, 1084.
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 229–244.
- Zhu, W.; Yu, J.; Nie, Y.; Shi, X.; Liu, Y.; Li, F.; Zhang, X.-l. Disequilibrium of M1 and M2 macrophages correlates with the development of experimental inflammatory bowel diseases. Immunol. Investig. 2014, 43, 638–652.
- Wang, W.; Li, X.; Zheng, D.; Zhang, D.; Peng, X.; Zhang, X.; Ai, F.; Wang, X.; Ma, J.; Xiong, W.; et al. Dynamic changes and functions of macrophages and M1/M2 subpopulations during ulcerative colitis-associated carcinogenesis in an AOM/DSS mouse model. Mol. Med. Rep. 2015, 11, 2397–2406.
- Lee, Y.S.; Song, S.J.; Hong, H.K.; Oh, B.Y.; Lee, W.Y.; Cho, Y.B. The FBW7-MCL-1 axis is key in M1 and M2 macrophage-related colon cancer cell progression: Validating the immunotherapeutic value of targeting PI3Kgamma. Exp. Mol. Med. 2020, 52, 815–831.
- Barbera-Guillem, E.; Nyhus, J.K.; Wolford, C.C.; Friece, C.R.; Sampsel, J.W. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res. 2002, 62, 7042–7049.
- Suarez-Lopez, L.; Sriram, G.; Kong, Y.W.; Morandell, S.; Merrick, K.A.; Hernandez, Y.; Haigis, K.M.; Yaffe, M.B. MK2 contributes to tumor progression by promoting M2 macrophage polarization and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E4236–E4244.
- Burmeister, K.; Quagliata, L.; Andreozzi, M.; Eppenberger-Castori, S.; Matter, M.S.; Perrina, V.; Grobholz, R.; Jochum, W.; Horber, D.; Moosmann, P.; et al. Vascular endothelial growth factor A amplification in colorectal cancer is associated with reduced M1 and M2 macrophages and diminished PD-1-expressing lymphocytes. PLoS ONE 2017, 12, e0175563.
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1β stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D 3. Oncogene 2009, 28, 3892–3902.
- Cai, J.; Xia, L.; Li, J.; Ni, S.; Song, H.; Wu, X. Tumor-associated macrophages derived TGF-β-induced epithelial to mesenchymal transition in colorectal cancer cells through smad2, 3-4/snail signaling pathway. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2019, 51, 252.
- Illemann, M.; Bird, N.; Majeed, A.; Sehested, M.; Laerum, O.D.; Lund, L.R.; Danø, K.; Nielsen, B.S. MMP-9 Is Differentially Expressed in Primary Human Colorectal Adenocarcinomas and Their Metastases. Mol. Cancer Res. 2006, 4, 293–302.
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64.
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 2016, 213, 2315–2331.
- Vu, T.; Datta, P.K. Regulation of EMT in Colorectal Cancer: A Culprit in Metastasis. Cancers 2017, 9, 171.
- Yun, J.A.; Kim, S.H.; Hong, H.K.; Yun, S.H.; Kim, H.C.; Chun, H.K.; Cho, Y.B.; Lee, W.Y. Loss of E-Cadherin expression is associated with a poor prognosis in stage III colorectal cancer. Oncology 2014, 86, 318–328.
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Xiong, B. M2 macrophages confer resistance to 5-fluorouracil in colorectal cancer through the activation of CCL22/PI3K/AKT signaling. OncoTargets Ther. 2019, 12, 3051.
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell. Physiol. Biochem. 2018, 45, 356–365.
- Herbeuval, J.-P.; Lelievre, E.; Lambert, C.; Dy, M.; Genin, C. Recruitment of STAT3 for production of IL-10 by colon carcinoma cells induced by macrophage-derived IL-6. J. Immunol. 2004, 172, 4630–4636.
- Cavnar, M.J.; Turcotte, S.; Katz, S.C.; Kuk, D.; Gönen, M.; Shia, J.; Allen, P.J.; Balachandran, V.P.; D’Angelica, M.I.; Kingham, T.P. Tumor-associated macrophage infiltration in colorectal cancer liver metastases is associated with better outcome. Ann. Surg. Oncol. 2017, 24, 1835–1842.
- Wang, D.; Wang, X.; Si, M.; Yang, J.; Sun, S.; Wu, H.; Cui, S.; Qu, X.; Yu, X. Exosome-encapsulated miRNAs contribute to CXCL12/CXCR4-induced liver metastasis of colorectal cancer by enhancing M2 polarization of macrophages. Cancer Lett. 2020, 474, 36–52.
- Wen, S.W.; Ager, E.I.; Christophi, C. Bimodal role of Kupffer cells during colorectal cancer liver metastasis. Cancer Biol. Ther. 2013, 14, 606–613.
- García-Pérez, R.; Fábrega, J.F.; Varona-Bosque, A.; Martínez, C.M.; Revilla-Nuin, B.; Cabellos, L.; Pena, R.; Vilana, R.; Gonzalez-Abós, C.; García-Valdecasas, J.C. Role of Kupffer cells in the progression of CRC liver metastases after the first stage of ALPPS. Sci. Rep. 2018, 8, 8089.
- Pereira, A.A.L.; Rego, J.; Morris, V.; Overman, M.J.; Eng, C.; Garrett, C.R.; Boutin, A.; Ferrarotto, R.; Lee, M.; Jiang, Z. Association between KRAS mutation and lung metastasis in advanced colorectal cancer. Br. J. Cancer 2015, 112, 424–428.
- Liu, H.; Liang, Z.; Zhou, C.; Zeng, Z.; Wang, F.; Hu, T.; He, X.; Wu, X.; Wu, X.; Lan, P. Mutant KRAS triggers functional reprogramming of tumor-associated macrophages in colorectal cancer. Signal Transduct. Target. Ther. 2021, 6, 144.
- Kuwahara, T.; Hazama, S.; Suzuki, N.; Yoshida, S.; Tomochika, S.; Nakagami, Y.; Matsui, H.; Shindo, Y.; Kanekiyo, S.; Tokumitsu, Y. Intratumoural-infiltrating CD4+ and FOXP3+ T cells as strong positive predictive markers for the prognosis of resectable colorectal cancer. Br. J. Cancer 2019, 121, 659–665.
- Ahrends, T.; Spanjaard, A.; Pilzecker, B.; Babala, N.; Bovens, A.; Xiao, Y.; Jacobs, H.; Borst, J. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 2017, 47, 848–861.
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271.
- De Simone, V.; Pallone, F.; Monteleone, G.; Stolfi, C. Role of TH17 cytokines in the control of colorectal cancer. Oncoimmunology 2013, 2, e26617.
- Kroemer, M.; Turco, C.; Spehner, L.; Viot, J.; Idirène, I.; Bouard, A.; Renaude, E.; Deschamps, M.; Godet, Y.; Adotévi, O. Investigation of the prognostic value of CD4 T cell subsets expanded from tumor-infiltrating lymphocytes of colorectal cancer liver metastases. J. Immunother. Cancer 2020, 8, e001478.
- Pedroza-Gonzalez, A.; Verhoef, C.; Ijzermans, J.N.; Peppelenbosch, M.P.; Kwekkeboom, J.; Verheij, J.; Janssen, H.L.; Sprengers, D. Activated tumor-infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology 2013, 57, 183–194.
- Katz, S.C.; Pillarisetty, V.; Bamboat, Z.M.; Shia, J.; Hedvat, C.; Gonen, M.; Jarnagin, W.; Fong, Y.; Blumgart, L.; D’Angelica, M. T cell infiltrate predicts long-term survival following resection of colorectal cancer liver metastases. Ann. Surg. Oncol. 2009, 16, 2524–2530.
- Katz, S.C.; Pillarisetty, V.G.; Bleier, J.I.; Kingham, T.P.; Chaudhry, U.I.; Shah, A.B.; DeMatteo, R.P. Conventional liver CD4 T cells are functionally distinct and suppressed by environmental factors. Hepatology 2005, 42, 293–300.
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102.
- Kobie, J.J.; Wu, R.S.; Kurt, R.A.; Lou, S.; Adelman, M.K.; Whitesell, L.J.; Ramanathapuram, L.V.; Arteaga, C.L.; Akporiaye, E.T. Transforming growth factor β inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003, 63, 1860–1864.
- Pryczynicz, A.; Cepowicz, D.; Zaręba, K.; Gryko, M.; Hołody-Zaręba, J.; Kędra, B.; Kemona, A.; Guzińska-Ustymowicz, K. Dysfunctions in the mature dendritic cells are associated with the presence of metastases of colorectal cancer in the surrounding lymph nodes. Gastroenterol. Res. Pract. 2016, 2016, 1–5.
- Gulubova, M.V.; Ananiev, J.R.; Vlaykova, T.I.; Yovchev, Y.; Tsoneva, V.; Manolova, I.M. Role of dendritic cells in progression and clinical outcome of colon cancer. Int. J. Color. Dis. 2012, 27, 159–169.
- Orsini, G.; Legitimo, A.; Failli, A.; Ferrari, P.; Nicolini, A.; Spisni, R.; Miccoli, P.; Consolini, R. Defective generation and maturation of dendritic cells from monocytes in colorectal cancer patients during the course of disease. Int. J. Mol. Sci. 2013, 14, 22022–22041.
- Nagorsen, D.; Voigt, S.; Berg, E.; Stein, H.; Thiel, E.; Loddenkemper, C. Tumor-infiltrating macrophages and dendritic cells in human colorectal cancer: Relation to local regulatory T cells, systemic T-cell response against tumor-associated antigens and survival. J. Transl. Med. 2007, 5, 62.
- Hsu, Y.-L.; Chen, Y.-J.; Chang, W.-A.; Jian, S.-F.; Fan, H.-L.; Wang, J.-Y.; Kuo, P.-L. Interaction between tumor-associated dendritic cells and colon cancer cells contributes to tumor progression via CXCL1. Int. J. Mol. Sci. 2018, 19, 2427.
- Legitimo, A.; Consolini, R.; Failli, A.; Orsini, G.; Spisni, R. Dendritic cell defects in the colorectal cancer. Hum. Vaccines Immunother. 2014, 10, 3224–3235.
- Xu, F.; Ye, Y.-J.; Liu, W.; Kong, M.; He, Y.; Wang, S. Dendritic cell/tumor hybrids enhances therapeutic efficacy against colorectal cancer liver metastasis in SCID mice. Scand. J. Gastroenterol. 2010, 45, 707–713.
- Miyagawa, S.; Soeda, J.; Takagi, S.; Miwa, S.; Ichikawa, E.; Noike, T. Prognostic significance of mature dendritic cells and factors associated with their accumulation in metastatic liver tumors from colorectal cancer. Hum. Pathol. 2004, 35, 1392–1396.
- Rodriguez, J.; Castañón, E.; Perez-Gracia, J.L.; Rodriguez, I.; Viudez, A.; Alfaro, C.; Oñate, C.; Perez, G.; Rotellar, F.; Inogés, S. A randomized phase II clinical trial of dendritic cell vaccination following complete resection of colon cancer liver metastasis. J. Immunother. Cancer 2018, 6, 96.
- Urosevic, J.; Garcia-Albéniz, X.; Planet, E.; Real, S.; Céspedes, M.V.; Guiu, M.; Fernandez, E.; Bellmunt, A.; Gawrzak, S.; Pavlovic, M. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat. Cell Biol. 2014, 16, 685–694.
- Zhao, S.; Jiang, T.; Zhang, L.; Yang, H.; Liu, X.; Jia, Y.; Zhou, C. Clinicopathological and prognostic significance of regulatory T cells in patients with non-small cell lung cancer: A systematic review with meta-analysis. Oncotarget 2016, 7, 36065.
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e91551.
- Merlo, A.; Casalini, P.; Carcangiu, M.L.; Malventano, C.; Triulzi, T.; Mènard, S.; Tagliabue, E.; Balsari, A. FOXP3 expression and overall survival in breast cancer. J. Clin. Oncol. 2009, 27, 1746–1752.
- Olguín, J.E.; Medina-Andrade, I.; Rodríguez, T.; Rodríguez-Sosa, M.; Terrazas, L.I. Relevance of regulatory T Cells during colorectal cancer development. Cancers 2020, 12, 1888.
- Ji, D.; Song, C.; Li, Y.; Xia, J.; Wu, Y.; Jia, J.; Cui, X.; Yu, S.; Gu, J. Combination of radiotherapy and suppression of Tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J. Immunother. Cancer 2020, 8, e000826.
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192.
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E. Two FOXP3+ CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684.
- Katz, S.C.; Bamboat, Z.M.; Maker, A.V.; Shia, J.; Pillarisetty, V.G.; Yopp, A.C.; Hedvat, C.V.; Gonen, M.; Jarnagin, W.R.; Fong, Y. Regulatory T cell infiltration predicts outcome following resection of colorectal cancer liver metastases. Ann. Surg. Oncol. 2013, 20, 946–955.
- Wang, L.; Hu, X.; Xu, Y.; Liu, Z. Arsenic trioxide inhibits lung metastasis of mouse colon cancer via reducing the infiltration of regulatory T cells. Tumour Biol. 2016, 37, 15165–15173.
- Huppert, L.A.; Green, M.D.; Kim, L.; Chow, C.; Leyfman, Y.; Daud, A.I.; Lee, J.C. Tissue-specific tregs in cancer metastasis: Opportunities for precision immunotherapy. Cell. Mol. Immunol. 2021, 1–13.
- Ellis, S.; Lin, E.J.; Tartar, D. Immunology of wound healing. Curr. Dermatol. Rep. 2018, 7, 350–358.
- Mazaki, J.; Katsumata, K.; Kasahara, K.; Tago, T.; Wada, T.; Kuwabara, H.; Enomoto, M.; Ishizaki, T.; Nagakawa, Y.; Tsuchida, A. Neutrophil-to-lymphocyte ratio is a prognostic factor for colon cancer: A propensity score analysis. BMC Cancer 2020, 20, 922.
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-β:“N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194.
- Itatani, Y.; Yamamoto, T.; Zhong, C.; Molinolo, A.A.; Ruppel, J.; Hegde, P.; Taketo, M.M.; Ferrara, N. Suppressing neutrophil-dependent angiogenesis abrogates resistance to anti-VEGF antibody in a genetic model of colorectal cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 21598–21608.
- Germann, M.; Zangger, N.; Sauvain, M.O.; Sempoux, C.; Bowler, A.D.; Wirapati, P.; Kandalaft, L.E.; Delorenzi, M.; Tejpar, S.; Coukos, G. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGF β. EMBO Mol. Med. 2020, 12, e10681.
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 983698.
- Gordon-Weeks, A.N.; Lim, S.Y.; Yuzhalin, A.E.; Jones, K.; Markelc, B.; Kim, K.J.; Buzzelli, J.N.; Fokas, E.; Cao, Y.; Smart, S. Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2–dependent angiogenesis in mice. Hepatology 2017, 65, 1920–1935.
- Yang, L.; Liu, L.; Zhang, R.; Hong, J.; Wang, Y.; Wang, J.; Zuo, J.; Zhang, J.; Chen, J.; Hao, H. IL-8 mediates a positive loop connecting increased neutrophil extracellular traps (NETs) and colorectal cancer liver metastasis. J. Cancer 2020, 11, 4384–4396.
- Yamamoto, T.; Kawada, K.; Itatani, Y.; Inamoto, S.; Okamura, R.; Iwamoto, M.; Miyamoto, E.; Chen-Yoshikawa, T.F.; Hirai, H.; Hasegawa, S.; et al. Loss of SMAD4 Promotes Lung Metastasis of Colorectal Cancer by Accumulation of CCR1+ Tumor-Associated Neutrophils through CCL15-CCR1 Axis. Clin. Cancer Res. 2017, 23, 833–844.
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-beta/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123.
- Lin, L.H.; Chang, K.W.; Cheng, H.W.; Liu, C.J. SMAD4 Somatic Mutations in Head and Neck Carcinoma Are Associated With Tumor Progression. Front. Oncol. 2019, 9, 1379.
- Liu, J.; Cho, S.N.; Akkanti, B.; Jin, N.; Mao, J.; Long, W.; Chen, T.; Zhang, Y.; Tang, X.; Wistub, I.; et al. ErbB2 Pathway Activation upon Smad4 Loss Promotes Lung Tumor Growth and Metastasis. Cell Rep. 2015, 10, 1599–1613.
- Jablonska, J.; Lang, S.; Sionov, R.V.; Granot, Z. The regulation of pre-metastatic niche formation by neutrophils. Oncotarget 2017, 8, 112132–112144.
- Wu, C.F.; Andzinski, L.; Kasnitz, N.; Kroger, A.; Klawonn, F.; Lienenklaus, S.; Weiss, S.; Jablonska, J. The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int. J. Cancer 2015, 137, 837–847.
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255.
- Glaire, M.A.; Domingo, E.; Sveen, A.; Bruun, J.; Nesbakken, A.; Nicholson, G.; Novelli, M.; Lawson, K.; Oukrif, D.; Kildal, W. Tumour-infiltrating CD8+ lymphocytes and colorectal cancer recurrence by tumour and nodal stage. Br. J. Cancer 2019, 121, 474–482.
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The immunoscore: Colon cancer and beyond. Clin. Cancer Res. 2020, 26, 332–339.
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139.
- O’Malley, G.; Treacy, O.; Lynch, K.; Naicker, S.D.; Leonard, N.A.; Lohan, P.; Dunne, P.D.; Ritter, T.; Egan, L.J.; Ryan, A.E. Stromal cell PD-L1 inhibits CD8+ T-cell antitumor immune responses and promotes colon cancer. Cancer Immunol. Res. 2018, 6, 1426–1441.
- Shan, T.; Chen, S.; Wu, T.; Yang, Y.; Li, S.; Chen, X. PD-L1 expression in colon cancer and its relationship with clinical prognosis. Int. J. Clin. Exp. Pathol. 2019, 12, 1764.
- Zhao, T.; Li, Y.; Zhang, J.; Zhang, B. PD-L1 expression increased by IFN-γ via JAK2-STAT1 signaling and predicts a poor survival in colorectal cancer. Oncol. Lett. 2020, 20, 1127–1134.
- Elfishawy, M.; Abd, E.S.A.; Hegazy, A.; El-Yasergy, D.F. Immunohistochemical Expression of Programmed Death Ligand-1 (PDL-1) in Colorectal carcinoma and Its Correlation with Stromal Tumor Infiltrating Lymphocytes. Asian Pac. J. Cancer Prev. 2020, 21, 225–232.
- FDA Approves First-Line Immunotherapy for Patients with MSI-H/dMMR Metastatic Colorectal Cancer. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-line-immunotherapy-patients-msi-hdmmr-metastatic-colorectal-cancer (accessed on 1 October 2021).
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380.
- Chang, L.-Y.; Lin, Y.-C.; Mahalingam, J.; Huang, C.-T.; Chen, T.-W.; Kang, C.-W.; Peng, H.-M.; Chu, Y.-Y.; Chiang, J.-M.; Dutta, A. Tumor-derived chemokine CCL5 enhances TGF-β–mediated killing of CD8+ T cells in colon cancer by T-regulatory cells. Cancer Res. 2012, 72, 1092–1102.
- Remark, R.; Alifano, M.; Cremer, I.; Lupo, A.; Dieu-Nosjean, M.-C.; Riquet, M.; Crozet, L.; Ouakrim, H.; Goc, J.; Cazes, A. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: Influence of tumor origin. Clin. Cancer Res. 2013, 19, 4079–4091.
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200.
- Li, Z.L.; Wang, Z.J.; Wei, G.H.; Yang, Y.; Wang, X.W. Changes in extracellular matrix in different stages of colorectal cancer and their effects on proliferation of cancer cells. World J. Gastrointest. Oncol. 2020, 12, 267–275.
- Yang, B.; Tang, F.; Zhang, B.; Zhao, Y.; Feng, J.; Rao, Z. Matrix metalloproteinase-9 overexpression is closely related to poor prognosis in patients with colon cancer. World J. Surg. Oncol. 2014, 12, 24.
- Dong, W.; Li, H.; Zhang, Y.; Yang, H.; Guo, M.; Li, L.; Liu, T. Matrix metalloproteinase 2 promotes cell growth and invasion in colorectal cancer. Acta Biochim. Biophys. Sin. 2011, 43, 840–848.
- Sis, B.; Sagol, O.; Kupelioglu, A.; Sokmen, S.; Terzi, C.; Fuzun, M.; Ozer, E.; Bishop, P. Prognostic significance of matrix metalloproteinase-2, cathepsin D, and tenascin-C expression in colorectal carcinoma. Pathol. Res. Prat. 2004, 200, 379–387.
- Zeng, Z.S.; Cohen, A.M.; Guillem, J.G. Loss of basement membrane type IV collagen is associated with increased expression of metalloproteinases 2 and 9 (MMP-2 and MMP-9) during human colorectal tumorigenesis. Carcinogenesis 1999, 20, 749–755.
- Sand, J.M.; Larsen, L.; Hogaboam, C.; Martinez, F.; Han, M.; Rossel Larsen, M.; Nawrocki, A.; Zheng, Q.; Karsdal, M.A.; Leeming, D.J. MMP mediated degradation of type IV collagen alpha 1 and alpha 3 chains reflects basement membrane remodeling in experimental and clinical fibrosis--validation of two novel biomarker assays. PLoS ONE 2013, 8, e84934.
- Brauchle, E.; Kasper, J.; Daum, R.; Schierbaum, N.; Falch, C.; Kirschniak, A.; Schaffer, T.E.; Schenke-Layland, K. Biomechanical and biomolecular characterization of extracellular matrix structures in human colon carcinomas. Matrix Biol. 2018, 68–69, 180–193.
- Bauer, J.; Emon, M.A.B.; Staudacher, J.J.; Thomas, A.L.; Zessner-Spitzenberg, J.; Mancinelli, G.; Krett, N.; Saif, M.T.; Jung, B. Increased stiffness of the tumor microenvironment in colon cancer stimulates cancer associated fibroblast-mediated prometastatic activin A signaling. Sci. Rep. 2020, 10, 50.
- Tanjore, H.; Kalluri, R. The role of type IV collagen and basement membranes in cancer progression and metastasis. Am. J. Pathol. 2006, 168, 715–717.
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518.
- Yuzhalin, A.E.; Gordon-Weeks, A.N.; Tognoli, M.L.; Jones, K.; Markelc, B.; Konietzny, R.; Fischer, R.; Muth, A.; O’Neill, E.; Thompson, P.R.; et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 2018, 9, 4783.
- Ganguly, D.; Chandra, R.; Karalis, J.; Teke, M.; Aguilera, T.; Maddipati, R.; Wachsmann, M.B.; Ghersi, D.; Siravegna, G.; Zeh, H.J. Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment. Cancers 2020, 12, 2652.
- Garvey, C.M.; Lau, R.; Sanchez, A.; Sun, R.X.; Fong, E.J.; Doche, M.E.; Chen, O.; Jusuf, A.; Lenz, H.-J.; Larson, B. Anti-EGFR Therapy Induces EGF Secretion by Cancer-Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance. Cancers 2020, 12, 1393.
- Chan, T.S.; Shaked, Y.; Tsai, K.K. Targeting the Interplay between Cancer Fibroblasts, Mesenchymal Stem Cells, and Cancer Stem Cells in Desmoplastic Cancers. Front. Oncol. 2019, 9, 688.
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013, 4, 1795.
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339.
- Rynne-Vidal, A.; Jimenez-Heffernan, J.A.; Fernandez-Chacon, C.; Lopez-Cabrera, M.; Sandoval, P. The Mesothelial Origin of Carcinoma Associated-Fibroblasts in Peritoneal Metastasis. Cancers 2015, 7, 1994–2011.
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718.
- Wang, L.; Yang, J.; Huang, J.; Wen, Z.-Q.; Xu, N.; Liu, X.; Zhang, J.-H.; Li, W.-L. MicroRNA expression profile in the N2 phenotype neutrophils of colorectal cancer and screen of putative key mircRNAs. Cancer Manag Res. 2020, 12, 5491–5503.
- Tauriello, D.V.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543.
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 10, 273.
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924.
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer 2014, 110, 724–732.
- You, J.; Li, M.; Cao, L.M.; Gu, Q.H.; Deng, P.B.; Tan, Y.; Hu, C.P. Snail1-dependent cancer-associated fibroblasts induce epithelial-mesenchymal transition in lung cancer cells via exosomes. QJM Int. J. Med. 2019, 112, 581–590.
- Xuefeng, X.; Hou, M.X.; Yang, Z.W.; Agudamu, A.; Wang, F.; Su, X.L.; Li, X.; Shi, L.; Terigele, T.; Bao, L.L.; et al. Epithelial-mesenchymal transition and metastasis of colon cancer cells induced by the FAK pathway in cancer-associated fibroblasts. J. Int. Med. Res. 2020, 48, 300060520931242.
- Shin, N.; Son, G.M.; Shin, D.H.; Kwon, M.S.; Park, B.S.; Kim, H.S.; Ryu, D.; Kang, C.D. Cancer-Associated Fibroblasts and Desmoplastic Reactions Related to Cancer Invasiveness in Patients With Colorectal Cancer. Ann. Coloproctol. 2019, 35, 36–46.
- Hawinkels, L.; Paauwe, M.; Verspaget, H.; Wiercinska, E.; Van Der Zon, J.; Van Der Ploeg, K.; Koelink, P.; Lindeman, J.; Mesker, W.; Ten Dijke, P. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107.
- Torres, S.; Garcia-Palmero, I.; Herrera, M.; Bartolome, R.A.; Pena, C.; Fernandez-Acenero, M.J.; Padilla, G.; Pelaez-Garcia, A.; Lopez-Lucendo, M.; Rodriguez-Merlo, R.; et al. LOXL2 Is Highly Expressed in Cancer-Associated Fibroblasts and Associates to Poor Colon Cancer Survival. Clin. Cancer Res. 2015, 21, 4892–4902.
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478.
- Suetsugu, A.; Osawa, Y.; Nagaki, M.; Saji, S.; Moriwaki, H.; Bouvet, M.; Hoffman, R.M. Imaging the recruitment of cancer-associated fibroblasts by liver-metastatic colon cancer. J. Cell. Biochem. 2011, 112, 949–953.
- Tsushima, H.; Ito, N.; Tamura, S.; Matsuda, Y.; Inada, M.; Yabuuchi, I.; Imai, Y.; Nagashima, R.; Misawa, H.; Takeda, H. Circulating transforming growth factor β1 as a predictor of liver metastasis after resection in colorectal cancer. Clin. Cancer Res. 2001, 7, 1258–1262.
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584.
- Zhang, R.; Qi, F.; Shao, S.; Li, G.; Feng, Y. Human colorectal cancer-derived carcinoma associated fibroblasts promote CD44-mediated adhesion of colorectal cancer cells to endothelial cells by secretion of HGF. Cancer Cell Int. 2019, 19, 192.
- Matsusue, R.; Kubo, H.; Hisamori, S.; Okoshi, K.; Takagi, H.; Hida, K.; Nakano, K.; Itami, A.; Kawada, K.; Nagayama, S. Hepatic stellate cells promote liver metastasis of colon cancer cells by the action of SDF-1/CXCR4 axis. Ann. Surg. Oncol. 2009, 16, 2645–2653.
- Mueller, L.; Goumas, F.A.; Affeldt, M.; Sandtner, S.; Gehling, U.M.; Brilloff, S.; Walter, J.; Karnatz, N.; Lamszus, K.; Rogiers, X. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am. J. Pathol. 2007, 171, 1608–1618.
- Kwak, Y.; Lee, H.E.; Kim, W.H.; Kim, D.-W.; Kang, S.-B.; Lee, H.S. The clinical implication of cancer-associated microvasculature and fibroblast in advanced colorectal cancer patients with synchronous or metachronous metastases. PLoS ONE 2014, 9, e91811.
- Schweiger, T.; Nikolowsky, C.; Starlinger, P.; Traxler, D.; Zimmermann, M.; Birner, P.; Hegedüs, B.; Dome, B.; Bergmann, M.; Mildner, M. Stromal expression of heat-shock protein 27 is associated with worse clinical outcome in patients with colorectal cancer lung metastases. PLoS ONE 2015, 10, e0120724.
- Bevacizumab. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125085s301lbl.pdf (accessed on 10 September 2021).
- Ramucirumab. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125477s034lbl.pdf (accessed on 10 September 2021).
- Chen, W.Z.; Jiang, J.X.; Yu, X.Y.; Xia, W.J.; Yu, P.X.; Wang, K.; Zhao, Z.Y.; Chen, Z.G. Endothelial cells in colorectal cancer. World J. Gastrointest. Oncol. 2019, 11, 946–956.
- Quiroz-Reyes, A.G.; Islas, J.F.; Delgado-Gonzalez, P.; Franco-Villarreal, H.; Garza-Trevino, E.N. Therapeutic Approaches for Metastases from Colorectal Cancer and Pancreatic Ductal Carcinoma. Pharmaceutics 2021, 13, 103.
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272.
- Zhang, D.; Bi, J.; Liang, Q.; Wang, S.; Zhang, L.; Han, F.; Li, S.; Qiu, B.; Fan, X.; Chen, W.; et al. VCAM1 Promotes Tumor Cell Invasion and Metastasis by Inducing EMT and Transendothelial Migration in Colorectal Cancer. Front. Oncol. 2020, 10, 1066.
- Cima, I.; Kong, S.L.; Sengupta, D.; Tan, I.B.; Phyo, W.M.; Lee, D.; Hu, M.; Iliescu, C.; Alexander, I.; Goh, W.L.; et al. Tumor-derived circulating endothelial cell clusters in colorectal cancer. Sci. Transl. Med. 2016, 8, 345ra389.
- Braet, F.; Nagatsuma, K.; Saito, M.; Soon, L.; Wisse, E.; Matsuura, T. The hepatic sinusoidal endothelial lining and colorectal liver metastases. World J. Gastroenterol. 2007, 13, 821–825.
- Zuo, Y.; Ren, S.; Wang, M.; Liu, B.; Yang, J.; Kuai, X.; Lin, C.; Zhao, D.; Tang, L.; He, F. Novel roles of liver sinusoidal endothelial cell lectin in colon carcinoma cell adhesion, migration and in-vivo metastasis to the liver. Gut 2013, 62, 1169–1178.
- Arteta, B.; Lasuen, N.; Lopategi, A.; Sveinbjornsson, B.; Smedsrod, B.; Vidal-Vanaclocha, F. Colon carcinoma cell interaction with liver sinusoidal endothelium inhibits organ-specific antitumor immunity through interleukin-1-induced mannose receptor in mice. Hepatology 2010, 51, 2172–2182.
- Zeng, X.; Ward, S.; Zhou, J.; Cheng, A. Liver Immune Microenvironment and Metastasis from Colorectal Cancer-Pathogenesis and Therapeutic Perspectives. Cancers 2021, 13, 2418.
- Gao, W.; Chen, L.; Ma, Z.; Du, Z.; Zhao, Z.; Hu, Z.; Li, Q. Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology 2013, 145, 636–646 e635.
- Tamura, M.; Oda, M.; Tsunezuka, Y.; Matsumoto, I.; Kawakami, K.; Watanabe, G. Vascular endothelial growth factor expression in metastatic pulmonary tumor from colorectal carcinoma: Utility as a prognostic factor. J. Thorac. Cardiovasc. Surg. 2004, 128, 517–522.
More