Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Renata Finelli and Version 2 by Amina Yu.
Heavy alcohol consumption (defined as more than 3 and 4 drinks in a day, or more than 7 and 14 drinks weekly, for women and men, respectively) is reported to negatively affect human health, promote traffic accidents, and alter social behaviors, with severe repercussions for personal, social, and professional lives. Clinically, alcohol consumption has been correlated with an increased incidence of different types of cancer , cardiovascular and liver diseases, birth defects, and psychiatric disorders.
- alcohol-related disorders
- ethanol
- ethyl alcohol
- infertility
- male
- spermatozoa
1. Ethanol-Induced Mechanisms of Cellular Damage
EtOH consumption has been reported to induce cellular damage through different interconnected mechanisms, which include the establishment of an inflammatory and oxidative environment, resulting in genotoxicity and enhanced apoptotic rate (Figure 2).
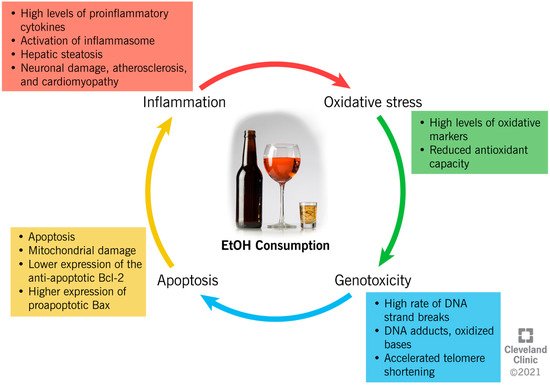
Figure 2. Ethanol-induced mechanisms of cellular damage.
The close link between chronic alcohol consumption and the onset of inflammation has been widely investigated [1][2][3][4][5][6][7][8][9][10][33,34,35,36,37,38,39,40,41,42]. In the liver, acetaldehyde activates inflammatory signaling pathways by increasing the synthesis of circulating proinflammatory cytokines by Kupffer cells [1][33], such as tumor necrosis factor-alpha (TNFα) [1][33]. TNFα acts on the hepatic stellate cells, further exacerbating the synthesis of proinflammatory and profibrogenic molecules, impairing cell function, and contributing to alcohol-induced cirrhosis [1][2][3][33,34,35]. In the liver, brain, and intestine, chronic alcohol administration has been proven to activate the inflammasomes. This is an intracytoplasmic multiprotein complex that induces the activation of caspase-1 and interleukin (IL)-1β, enhancing the proinflammatory status in the same tissues [4][5][36,37]. Moreover, through a mechanism of negative feedback, inflammation enhances the negative impact of alcohol metabolism through the reduced expression of ADH1 and ALDH2, which causes systemic and hepatic increases in the EtOH and acetaldehyde levels [8][40]. In addition, the metabolism of acetaldehyde into acetate increases the NADH:NAD+ ratio within the cell, and consequently inhibits the fatty acid β-oxidation pathway in the liver. This results in the accumulation of triglycerides in the hepatocytes as hepatic steatosis, and the enhancement of inflammation [6][38].
EtOH exposure alters the NF-kB pathway, which is important in inflammatory and immune responses in the brain, and decreases the transcription of regulatory cyclic protein AMP-responsive element-binding protein (CREB), affecting neuronal survival and protection from apoptosis [10][42].
Moreover, alcohol consumption can increase oxidative stress through different mechanisms, with serious consequences for human health. However, ROS are highly unstable and are reactive with proteins, lipids, and any cellular structures. When ROS are overproduced, they switch the cellular redox status towards oxidative stress, which contributes to several pathological conditions [11][44].
EtOH metabolism enhances ROS generation, as the activity of dehydrogenases and the CYP2E1 system increases the levels of NADH, which, in turn, is oxidized by xanthine oxidase, with the generation of ROS [12][45]. An increased oxidative status is also due to the increased peroxisomal activity in the livers of heavy drinkers, as a result of steatosis [13][46].
In addition to directly increasing the production of ROS, in vitro studies have shown that acetaldehyde reportedly causes an intracellular redox imbalance by increasing the levels of oxidative stress markers (i.e., malondialdehyde (MDA)), and by reducing the concentration of antioxidant glutathione (GSH) [14][15][47,48]. Acetaldehyde also impairs the enzymatic activity of superoxide dismutase (SOD) 2, a powerful endogenous antioxidant [16][49]. In alveolar macrophages, chronic alcohol consumption and the subsequent onset of oxidative stress alter the expression of the NADPH oxidases (Noxes), enzymes contributing to the phagocyte-mediated host defense [17][50]. EtOH consumption upregulates the expression of the Noxes, as well as the regulatory proteins, p22phox, p47phox, and p67phox, both in vivo and in vitro, resulting in the higher intracellular production of O₂− and H2O2, and lower GSH levels in the lung tissue [17][50].
The establishment of an inflammatory and oxidative environment modifies cellular homeostasis and DNA integrity, and can have a genotoxic/mutagenic impact, eventually leading to cell death. In vitro and in vivo studies indicate that EtOH induces autophagy in neurons by inhibiting the expression of the antiapoptotic Bcl-2 family proteins, while it increases the expression of proapoptotic proteins [18][51]. The expression of Bcl-2 is also modulated by EtOH through epigenetic regulation. In fact, by increasing the histone deacetylase activity, EtOH affects the binding of acetyl-histone H3 to the Bcl-2 promoter, which results in a reduced Bcl-2 gene transcription [19][52]. Furthermore, it has been suggested that EtOH induces programmed cell death through the mitochondrial pathway, as the inhibition of caspase-9 (key molecule in this pathway) resulted in a reduced apoptotic rate [20][53]. The activity of caspase-3, an apoptotic marker, was also increased in the cerebral cortices of rats after EtOH treatment [21][54], possibly because of the depolarization of the mitochondrial membrane by acetaldehyde accumulation [22][55]. The role of alcohol metabolites as mediators of mitochondrial damage was also demonstrated in knock-out mice for ALDH or ADH enzymes, which showed reduced membrane potential in comparison to the controls [22][55].
However, whereas evidence shows a negative impact of high-dose EtOH on cellular physiology, a low dose seems to reduce the apoptotic rate. In fact, recent data suggests that a low dose of EtOH contributes to the regulation of the expression of the TNF receptor, p75NTR, and a potassium-chloride transporter (KCC2) [23][56]. While p75NTR is involved in the regulation of apoptosis, KCC2 regulates the cellular electrochemical gradient, and its expression is crucial to maintaining the classic hyperpolarizing GABAergic inhibition in mature adult neurons [23][56].
2. Alcohol Consumption and Male Infertility: Evidence from Animal and Human Studies
2.1. Impact of Alcohol on Reproductive Hormonal Regulation
The secretion of reproductive hormones, such as testosterone, is regulated by the hypothalamic–pituitary–gonadal (HPG) axis [24][114]. The hypothalamus releases gonadotropin-releasing hormone (GnRH), which reaches the adenohypophysis through the hypophyseal portal system. As a response, the GnRH triggers the release of gonadotropins (luteinizing hormone (LH), and follicle-stimulating hormone (FSH)), which act at the testicular level. LH stimulates the Leydig cells to produce testosterone [24][114]. LH binding triggers the internalization of cholesterol, which is converted to pregnenolone, and then to 17α-hydroxy-pregnenolone, and androgen dehydroepiandrosterone (DHEA). DHEA is converted to androstenedione via 3β-hydroxysteroid dehydrogenase (3β-HSD), and then to testosterone via 17β-hydroxysteroid dehydrogenase-3 (17β-HSD-3) [25][115]. FSH binds on the Sertoli cells, supporting the spermatogenesis. This process includes three phases of development: (1) The proliferation of spermatogonial cellular precursors (mitosis); (2) Maturation into spermatocytes, with recombination, reduction, and division of DNA (meiosis); and (3) Differentiation into spermatids, and, finally, into mature spermatozoa (spermiogenesis) [26][116].
Alcohol consumption seems to affect endocrine functions, compromising the regulation of the HPG axis; however, contradictory evidence is reported in the literature. In 2005, a study investigating fertility hormones in 66 heavy drinkers reported increased levels of FSH, LH, and estradiol [27][79]. Conversely, Maneesh et al. reported lower levels of the gonadotropins, FSH and LH, in 45 alcoholic men [28][80], while Jensen et al. did not observe any association between the levels of FSH, LH, and inhibin B and the intake of alcohol in 8344 healthy men [29][81]. Similar results are reported regarding testosterone concentration, with studies observing higher values in alcoholic men [29][30][31][81,82,83], while others observed lower testosterone [27][28][79,80]. In addition, chronic alcohol intake can increase serum prolactin (hyperprolactinemia), causing hypogonadism, reduced sperm production, impotence, and gynecomastia in men [32][33][117,118]. These differences could be due to the heterogeneity in the study designs, the populations included, and the different approaches to classifying drinking habits, whereas the studies conducted in animal models reported a reduction in the circulating levels of LH and FSH following EtOH exposure [34][35][36][37][38][39][67,68,69,70,71,72].
Studies dating back to the 1980s suggest that acute EtOH intake might directly inhibit the hypothalamic release of GnRH, and, consequently, reduce the levels of LH and testosterone [40][41][119,120], probably through the alteration of the nitric oxide (NO) pathway. NO stimulates the synthesis of PGE2 in tissues by binding the heme group of cyclooxygenase-1 [42][121], while EtOH inhibits the cyclooxygenase-1 activity, and eventually the synthesis of PGE2 and GnRH secretion [43][122]. Alcohol consumption has also been associated with a reduction in the number of Leydig cells and an altered morphology [44][78]. The Leydig cells of rats fed with EtOH showed reduced sizes, swollen mitochondria, and smaller amounts of cytosol, along with the reduced synthesis of testosterone [44][78]. Besides altering its synthesis, alcohol consumption is also associated with an enhanced rate of testosterone elimination [45][46][17,73]. In fact, EtOH stimulates the activity of aromatase, which converts testosterone into estradiol [46][73], resulting in elevated estrogen levels and abnormal breast enlargement, as observed in heavy alcoholics [45][31][17,83]. This further inhibits the synthesis of FSH and LH, and, consequently, the synthesis of testosterone itself [47][48][74,75]. Moreover, chronic alcohol intake has been associated with the reduced bioavailability of insulin growth factor (IGF)-1, which physiologically stimulates the synthesis of testosterone [49][76]. Finally, it is important to mention that NAD is the cofactor of enzymes involved in both EtOH and androgen metabolism. Therefore, high EtOH intake reduces NAD+/NADH levels, and indirectly inhibits the activity of enzymes involved in testosterone synthesis [50][77].
2.2. Impact of Alcohol Consumption on Semen Quality
Data from an animal study showed that an EtOH-rich diet can affect testicular function, with consequences on the semen quality. In fact, EtOH-fed mice showed compromised integrities of the testis and seminal vesicles, and altered weight of the prostate, which resulted in increased germ cell desquamation, decreased sperm concentrations, and increased abnormal sperm morphologies [51][123]. Besides the alterations in the semen quality (lower sperm concentration, motility, and percentage of normal forms), Rahimipour et al. also reported reduced DNA condensation and integrity in mice fed with ethanol compared to controls, along with increased apoptotic rates [52][85]. In addition, in vitro experiments showed an accelerated acrosomal loss occurring during the sperm capacitation of human and animal sperm incubated in ethanol, further reducing their fertilizing ability [53][54][55][56][124,125,126,127]. This is probably due to the capacity of ethanol to alter lipid fluidity and membrane permeability through the oxidation of the membranes’ lipids and proteins [56][127]. In rats, decreased sperm motility was observed after exposure to EtOH, as well as changes in the meiotic divisions, reduced gametes viability, and a higher rate of sperm with poorly condensed chromatin [57][58][86,87]. In humans, a case study reported severe oligoasthenoteratozoospermia in an alcoholic man, which evolved into cryptozoospermia, and then azoospermia after a few years [59][91]. In 2017, a meta-analysis investigated the impact of alcohol intake on semen quality by analyzing evidence from 18 cross-sectional studies [60][93]. The authors concluded that daily alcohol consumption results in a worsened semen quality, particularly in terms of the semen volume and the sperm morphology. However, this effect was not reported for occasional drinkers, while the authors observed even better sperm motility in occasional drinkers than never drinkers, despite all the limitations identified in their analysis [60][93]. In fact, the association between semen quality and the amount of alcohol consumed is still controversial. Surprisingly, Ricci et al. observed a positive correlation between semen volume and concentration, and moderate alcohol consumption (equal to 4–7 units/week), suggesting that a limited consumption of alcohol may improve semen quality [61][96]. This might be explained by the fact that some compounds present in alcoholics drinks (i.e., natural flavonoids, and polyphenols in red wine) have antioxidant and anti-inflammatory activities, and they reportedly have a positive influence on semen quality (particularly by improving sperm motility, concentration, and survival) at low concentrations [62][63][64][65][128,129,130,131]. However, a cross-sectional study including 8344 healthy men did not report any association between low/moderate alcohol consumption and semen quality [29][81]. Similarly, other studies failed to identify any coherent dose–response pattern in the semen parameters depending on the degree of alcohol consumption [31][66][67][68][69][83,88,132,133,134]. Boeri et al. suggested that the correlation between alcohol consumption and alterations in the semen parameters might be directly proportional to the amount of alcohol consumed. In fact, the semen parameters were reportedly worse in samples of heavy rather than moderate drinkers [70][89]. Several recent studies of different global geographic regions have confirmed the negative impact of heavy alcohol consumption on semen quality. In fact, in China, a cross-sectional study conducted in 2020 reported reduced sperm concentrations in 55 heavy drinkers suffering from secondary infertility [66][88], while in Italy, 45 heavy drinkers with primary infertility showed reduced sperm concentrations and motilities compared to moderate drinkers or abstainers [70][89]. Similarly, an inverse association between sperm counts and alcohol consumption was observed in a Brazilian population of 167 infertile men [71][90], while a large study conducted on a Danish population of 1221 men showed a direct association between worsening semen quality and increasing alcohol intake [72][92]. Other studies have also confirmed a higher rate of sperm DNA fragmentation and chromatin decondensation in heavy drinkers [70][71][73][74][89,90,94,95].
The differences in the study designs, and the discrepancies in the published studies, make it challenging to draw any conclusions regarding the association between the amount of alcohol consumed and the semen quality. Hence, much research is still warranted in this regard.
2.3. Impact of Alcohol Consumption on Gene Transcription, Genetics, and Epigenetics Regulation
Male infertility associated with chronic alcohol consumption might also be due to a differential regulation of gene expression, followed by an altered metabolism of the specific proteins involved in sperm maturation [75][76][77][78][97,98,101,135]. In fact, it was shown that EtOH can lead to oxidative damage in the epididymis by altering the mRNA expression of β-defensin, which has antimicrobial properties and is involved in sperm function [75][97]. Alcohol exposure also altered post-transcriptional RNA modifications in murine sperm, thereby influencing the expression of small mitochondrial RNA species, the mitochondrial function of spermatozoa, and the reproductive ability [77][101]. In addition, EtOH compromised sperm viability by reducing the expression of proliferating cell nuclear antigen (PCNA) in germ cells and by promoting apoptosis [78][135].
Several genetic variants of enzymes involved in EtOH metabolism have been identified, associated with different degrees of tolerability to alcohol. In fact, specific single nucleotide polymorphisms in ADH and ALDH show altered enzymatic activity, which results in the accumulation of acetaldehyde. Because of its unpleasant effects, this is associated with a lower risk of developing an alcohol-use disorder [77][101].
A correlation between EtOH consumption and epigenetic changes in sperm DNA has been explored in various studies [79][77][80][81][82][100,101,102,103,136]. In this regard, aberrant gene methylation in sperm DNA was associated with male infertility [80][102]. During early embryonic development, there are two loci in paternal DNA (differentially methylated region upstream of the H19 gene-H19 DMR, and intergenic differentially methylated region, IG-DMR) that are highly methylated, and that are fundamental for growth and neurobehavioral development [81][103]. A significant difference was observed in the demethylation rates of specific C-phosphate-G (CpG) sites between nondrinkers and moderate drinkers. Particularly, there was a direct correlation between the amount of alcohol consumed and the degree of demethylation at the H19 DMR and IG-DMR genes, supporting the hypothesis that EtOH consumption may reduce the DNA methyltransferase activity, and increase the risk of the transmission of defective imprinted genes [81][103]. In addition, as shown in the rat testis, EtOH exposure may affect spermatogenesis, as it increases the acetylation of lysine 9 in histone 3 (Ac-H3-lys9), which results in impaired sperm chromatin organization and embryonic development [79][100].
2.4. Consequences of Paternal Alcohol Consumption on the Offspring
Several studies were conducted in animal models to investigate the transgenerational effect of paternal alcohol exposure, showing low fetal and birth weights in the offspring, as well as altered organ weights and hormonal regulations (for a review on the topic, see [77][101]). An animal study showed that paternal alcohol exposure led to hormonal and nervous system anomalies in the offspring. Specifically, the expression of nerve growth factor (NGF), a well-characterized neurotrophin involved in the development of the nervous system in vertebrates [83][105], was strongly reduced in the frontal cortices of the offspring of EtOH-fed mice, as well as in the hippocampal, hypothalamic, and olfactory lobes, leading to the conclusion that paternal alcohol consumption can produce critical alterations in the brains of offspring [84][106]. The anogenital distance, which is a male fertility marker related to the proper functioning of the endocrine system, was also shorter in the offspring of EtOH-consuming fathers, indicating that alcohol may have an adverse effect on the reproductive development of offspring [85][107]. Paternal alcoholism before conception was also associated with limited offspring growth and decreased placental efficiency [86][104].
In a case of paternal alcohol exposure, the offspring showed reproductive dysfunctions similar to those reported for direct alcohol consumption, including alterations in the hormonal axis and the semen quality [87][113].
Three possible mechanisms have been proposed to explain the effect of paternal alcohol consumption on the offspring: (a) An alteration of the sperm chemical composition, leading to behavioral, biochemical, and hormonal disturbances in the offspring; (b) A failure of the elimination of EtOH-damaged sperm; and (c) An induction of genetic mutations in sperm DNA that can be transmitted to the offspring [88][137]. Although chemical alterations in sperm and/or seminal plasma may compromise embryonic development, this remains to be confirmed. The second hypothesis refers to the mechanisms of natural selection that may fail to remove spermatozoa damaged by EtOH during spermatogenesis, in favor of genetically intact spermatozoa. The third hypothesis emphasizes a heritable genetic change in EtOH consumers, and seems to be the most accepted [88][137]. However, a study published in 2017 was not able to identify any association between the phenotypes observed in mice exposed to EtOH (fetal growth restriction and altered developmental programming) and the paternal DNA methylation profiles, questioning the transgenerational effect of EtOH exposure [89][138]. EtOH-related epigenetic effects on the paternal germline might provide an explanation for the transgenerational influence of the father’s lifestyle habits on the development of the offspring, and it surely deserves more investigation [90][139].