Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Shanel Raghubeer and Version 3 by Jason Zhu.
The 5-10-methylenetetrahydrofolate reductase (MTHFR) enzyme is vital for cellular homeostasis due to its key functions in the one-carbon cycle, which include methionine and folate metabolism, and protein, DNA, and RNA synthesis.
- Methylenetetrahydrofolate
- Inflammation
- Vitamin B
1. Introduction
5-10-Methylenetetrahydrofolate reductase (MTHFR) is an essential enzyme in folate and homocysteine (Hcy) metabolism. The gene for this enzyme is located on the short arm of chromosome 1 (1p36.3) and encodes for dimeric proteins [1]. The major product of the MTHFR gene is a 77 kDa protein, with a second 70 kDa isoform found in humans [2]. MTHFR catalyses the irreversible reduction of 5-10-MTHF to 5-methylTHF, a circulatory form of folate used in the remethylation of Hcy to methionine [2][3][2,3]. In total, 34 rare but deleterious mutations have been reported, and the location of the mutation may influence its impact [4]. For example, relative to their size, exons 5 and 6 harbour the most mutations, which may be more deleterious since these exons are located within the catalytic domain and may play a role in substrate binding [4]. Mudd et al. published the first report on MTHFR involvement in disease upon identifying a patient with homocystinuria due to severe MTHFR deficiency [5]. In 1988, a thermolabile variant of the MTHFR enzyme was identified in patients with cardiovascular disease (CVD). This deficiency was milder but more common and induced mild elevation of Hcy levels, which may be an independent risk factor for CVD [6].
The most common cause of elevated Hcy levels is thought to be the C677T MTHFR polymorphism (rs 1801133). This polymorphism involves the substitution of cytosine with thymine at position 677, resulting in an amino acid change from alanine to valine in the enzyme [1][7][8][1,7,8]. This common MTHFR gene mutation affects Hcy levels and is thought to contribute to hyperhomocysteinemia, reduced folate levels, and several CVD-associated diseases [9]. The C677T mutation reportedly disrupts thermostability, leading to enzyme dysfunction [10]. This product is termed thermolabile since enzyme activity is reduced at temperatures above 37 °C [11]. MTHFR enzyme activity appeared 50–60% lower in C677T homozygous (TT) patients, with activity decreasing by 65% at 46 °C [10]. Another commonly encountered polymorphism occurs at position 1298 on the MTHFR gene and involves the substitution of adenine with cytosine, resulting in an amino acid change from glutamate to alanine in the enzyme [12]. This mutation also produces reduced enzyme activity although not to the same extent as that caused by the C677T polymorphism [12].
2. MTHFR in Inflammation
Inflammation is mediated primarily by the nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) pathway, which involves several related transcription factors. The NF-κB protein is sequestered and rendered inactive in the cytoplasm during normal conditions by the inhibitor of nuclear factor kappa B alpha (IκBα) protein [13][17]. NF-κB is activated by certain signals, such as reactive oxygen species (ROS) and pro-inflammatory cytokines (interleukin (IL)-1, IL-6, and tumour necrosis factor alpha (TNFα)), and translocates into the nucleus to activate the inflammatory cascade, thus regulating the inflammatory response [13][14][17,18]. Inflammation occurs to remove potentially harmful stimuli, such as pathogens, toxins, or damaged cells, and promote tissue homeostasis [15][19]. However, if acute inflammation is not resolved, chronic inflammation, which is associated with the development and progression of numerous illnesses, may occur [15][19]. C-reactive protein (CRP) is an acute-phase protein primarily synthesised by hepatocytes and to a lesser extent by smooth muscle cells, lymphocytes, and adipocytes [16][20]. Increasing evidence indicates that CRP plays a role in inflammatory responses and other immune system responses, such as apoptosis and the production of cytokines, especially TNFα and IL-6. Further, CRP has been shown to increase up to 1000-fold at infection or inflammation sites and has been associated with CVDs [16][20]. Several factors can influence CRP levels, including weight, lipid profile, age, gender, and smoking status, as well as polymorphisms in the CRP gene [17][21]. Diabetes patients with the C677T polymorphism have reportedly exhibited increased levels of CRP and IL-6 [18][22].
Regarding the MTHFR C677T polymorphism and inflammation, Dedoussis et al. reported an association between the homozygous mutant genotype (TT) and increased inflammatory markers [19][23]. The study investigated inflammatory markers and MTHFR C677T genotype association in 574 CVD-free subjects from the Attica region in Greece and reported that 11% of subjects presented with the homozygous TT genotype, 48% with the heterozygous CT genotype, and 41% with the homozygous CC genotype (normal). Inflammatory markers, including CRP, white blood cell counts, and amyloid-A levels, appeared elevated in the TT genotype subjects compared to the CC and CT genotype individuals despite controlling for confounding variables [19][23]. Mild MTHFR deficiency caused by the C677T polymorphism was found to affect inflammatory and lipid pathways in a murine model [20][24]. Researchers found that MTHFR deficiency reduced s-adenosylmethionine (SAM) and betaine levels and increased s-adenosylhomocysteine (SAH) levels, resulting in reduced methylation capacity and alteration of inflammatory mediators. Furthermore, MTHFR-deficient mice fed a high-fat diet exhibited exacerbated effects in inflammatory and lipid pathways, with increased inflammation and lipid accumulation. These results suggest that MTHFR deficiency coupled with a low-quality high-fat diet may increase the risk of developing non-alcoholic fatty liver disease (NAFLD) due to disrupted methylation capacity, lipid metabolism, and inflammatory responses [20][24]. An estimated one billion people have NAFLD [21][25], which is characterised by inflammation and ectopic fat in the liver, causing liver dysfunction, fibrosis, end-stage liver disease, and cirrhosis [22][26]. Research has provided evidence highlighting the influence of microRNAs (miRNA) on NAFLD [23][24][25][26][27,28,29,30]. MicroRNAs are short, non-coding RNA molecules that function to regulate the expression of messenger RNA (mRNA), thereby influencing gene expression and protein synthesis [22][26]. An et al. investigated the effect of the C677T MTHFR polymorphism and miR-149 on NAFLD. Results showed that miR-149 was upregulated in TT genotyped hepatocytes treated with long-chain fatty acid (FFA), while MTHFR was downregulated in these hepatocytes [22][26]. However, FFA did not affect hepatocytes genotyped as CC, suggesting that the polymorphism influences the response to FFA and the development of NAFLD. miR-149, a regulator of the MTHFR gene, was upregulated in response to FFA in TT genotyped hepatocytes and in turn decreased MTHFR expression [22][26]. This study suggests that MTHFR genotype affects susceptibility to NAFLD.
The MTHFR C677T polymorphism was shown to be associated with inflammatory diseases, such as psoriasis (Caucasian Czech population) [27][31] and inflammatory bowel disease (Caucasian Irish population) [28][32], while both C677T and A1298C variants have been linked to rheumatoid arthritis in a Caucasian and Asian population [29][30][33,34]. Systemic inflammation has been linked with several diseases, including diabetes, coronary artery disease, and cancer. Studies have shown the presence of inflammatory processes in pancreatic beta cells of diabetes patients [31][35], and insulin resistance has been linked to cytokine-induced inflammation [32][36]. Furthermore, inflammatory cells and cytokines (TNFα, IL-1, IL-6, and monocyte chemoattractant protein-1 (MCP-1)) are thought to play important roles in the development and progression of atherosclerosis [33][37]. Khalighi et al. investigated whether MTHFR gene polymorphisms contributed to systemic inflammation in a cohort of 292 patients by determining two markers of systemic inflammation, namely neutrophil-to-lymphocyte ratio (NLR) and platelet-to-lymphocyte ratio (PLR) [33][37]. This study reported that the A1298C variant produced opposite effects compared to the C677T variant. Results showed that patients with the C677T variant displayed a greater NLR than C677T wild-type patients, while those with the A1298C variant displayed lower NLR and PLR than A1298C wild-type patients [33][37]. The above-mentioned studies provide compelling evidence that suggests a link between systemic inflammation and MTHFR gene polymorphisms.
3. Vitamin B Pathway and MTHFR Effects
B vitamins play important roles in the one-carbon cycle (Figure 1), which is essential for cellular function. The one-carbon cycle consists of interlinking biochemical pathways that include folate and methionine metabolism [34][38]. These metabolic pathways generate methyl groups for several essential processes, including DNA synthesis, antioxidant generation, and amino acid homeostasis [35][39]. Furthermore, methyl groups are involved in methylation reactions in epigenetic regulation, thereby influencing gene expression and protein synthesis [36][40]. One-carbon metabolism is regulated by methionine, vitamin B12, and vitamin B9 (folate), which are essential dietary requirements [35][39]. Folate and methionine cycles are linked by a rate-limiting enzyme, methionine synthase (MS), which converts Hcy to methionine using 5-methylTHF (produced by MTHFR) and B12 [37][41]. Ultimately, this cycle produces methyl donors that participate in essential cellular processes [38][42]. Therefore, genetic polymorphisms and vitamin B deficiencies can be detrimental to the one-carbon cycle, which may result in Hcy accumulation and vascular endothelium damage [39][43].
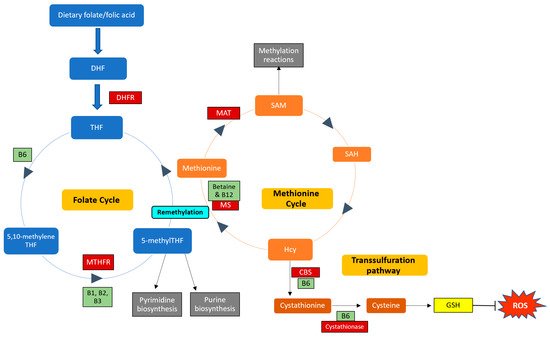
Figure 1. Enzymes and cofactors involved in the one-carbon cycle, including methionine and folate metabolism. Dietary folate (vitamin B9) is converted into dihydrofolate (DHF) by the enzyme dihydrofolate reductase (DHFR) and is then reduced to tetrahydrofolate (THF). THF is converted to 5,10-methyleneTHF, which is converted to 5-methylTHF by the MTHFR enzyme (using vitamins B1, B2, and B3 as cofactors). 5-methylTHF is then used as a methyl donor in pyrimidine and purine synthesis and can donate a methyl group to regenerate methionine from homocysteine (Hcy); this reaction is catalysed by methionine synthase (MS) with vitamin B12 as a cofactor. This is termed remethylation. Dietary betaine from the liver can also serve as a methyl donor and participate in remethylation. Thereafter, methionine adenosyltransferase (MAT) catalyses the transfer of adenosine to methionine to generate s-adenosylmethionine (SAM), which functions in methylation reactions. SAM is then demethylated and forms s-adenosylhomocysteine (SAH), which is hydrolysed to form Hcy. Hcy can now enter the transsulfuration pathway to form cystathionine (catalysed by cystathionine beta-synthase (CBS)) and cysteine (catalysed by cystathionase). Cysteine is then used to synthesise glutathione (GSH), thereby regenerating antioxidant levels to combat damage by reactive oxygen species (ROS). Enzymes and cofactors are indicated in dark red and light green squares, respectively.
The C677T and A1298C polymorphisms are thought to be primary contributors to hyperhomocysteinemia, resulting in an increased risk of developing thrombosis [40][44]. Xuan et al. reported that the C677T polymorphism is associated with a 50% reduction in MTHFR enzyme activity as well as increased plasma Hcy and reduced plasma folic acid concentrations, which may promote endothelial dysfunction and increase the risk of developing various CVDs [41][45]. Characteristics of MTHFR enzyme dysfunction include elevated plasma Hcy levels, which is often used as the first indicator, increased plasma SAH and cystathionine, and decreased methionine and SAM [3]. These imbalances are attributed to dysfunctional methionine metabolism, which may further influence DNA methylation and gene expression. MTHFR-deficient patients may present with developmental delays, neurological and vascular issues, seizures, or thrombosis [2]. Severe MTHFR deficiencies are rare but are also the most commonly encountered inborn errors of folate metabolism [2].
Research has reported an association between hyperhomocysteinemia and low bone mineral density (BMD) as well as CRP, vitamin D, vitamin B12, and folate levels in postmenopausal women [42][46]. Two hundred and fifty women (aged 50–65 years) were recruited and divided into two groups based on BMD. Of the 250 women, 155 had low BMD, and 97 women were within the normal BMD range. Women with low BMD presented with increased Hcy and CRP levels and decreased levels of vitamin B12, vitamin D, and folate [42][46]. Furthermore, 77% of women with low BMD presented with the C677T MTHFR polymorphism, while only 37% of women with normal BMD presented with the mutation. Taken together, these results suggest an association between the C677T MTHFR polymorphism and elevated Hcy levels and inflammation, which may further influence osteoporosis onset [42][46]. The MTHFR enzyme is involved in the formation of SAM, which donates a methyl group for the progression of reactions catalysed by catechol-O-methyltransferase (COMT), an enzyme that participates in hormone regulation, thereby inactivating catechol oestrogens and playing a role in oestrogen metabolism [43][47]. Catechol oestrogens may contribute to increased ROS production, thereby inducing DNA damage, oxidative cell damage, and inflammation [44][48], and may further influence carcinogenesis and the development of CVDs and related diseases [45][46][49,50]. Research has shown that oestrogen-induced hypertension is age- and sex-specific; therefore, polymorphisms in the MTHFR gene may contribute to disrupted oestrogen metabolism and in turn influence oestrogen-associated diseases [45][49].
Vitamin B12 deficiency is thought to contribute to increased Hcy levels. Patients with low folic acid levels often present with low levels of B12 and other B vitamins [47][51]. In a study investigating the relationship between folate and homocysteine levels, sub-normal folate levels (<2 ng/mL) resulted in two-fold increases in Hcy serum levels, while low folate levels (2–3.9 ng/mL) resulted in increased Hcy levels compared to patients with normal folate levels (5–17.9 ng/mL) [47][51]. Therefore, folic acid and vitamin B12 supplementation is thought to improve hyperhomocysteinemia. Several studies have reported higher Hcy levels in patients with the TT genotype compared to CC, often coupled with low plasma folic acid levels [48][49][50][52,53,54]. Pereira et al. reported that plasma Hcy levels appeared increased in TT vs. CC patients (16.2 vs. 8.2 µmol/L), while low vitamin B12 levels were observed in TT vs. CC patients (196 vs. 301 pmol/L) [51][55]. Moreover, folate deficiency results in thymidine depletion, which increases the incorporation of uracil into DNA, resulting in impaired DNA repair and an increased likelihood of malignant transformation due to DNA instability and chromosome aberrations [52][56].