Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ahmed N. Al-Sabi and Version 2 by Peter Tang.
Voltage-gated K+ (Kv) channels are intrinsic plasma membrane proteins mediating the selective flow of potassium ions in response to depolarization of the transmembrane electric field. Their ionic selectivity and voltage dependence allow Kv channels to be central players in virtually all physiological functions, including the maintenance and modulation of neuronal and muscular (both cardiac and skeletal) excitability, regulation of calcium signaling cascades, control of cell volume, immune response, hormonal secretion, and others.
- bioactives
- Kv1
- Conotoxins
- Potassium channel modulators
- peptide neurotoxins
1. Kv1 Channels
Voltage-gated K+ (Kv) channels are intrinsic plasma membrane proteins mediating the selective flow of K+ ions down their electrochemical gradient in response to depolarization of the cell's transmembrane electric field [1]. The selectivity and voltage dependence of Kv channels make them central players in virtually all physiological functions, including the maintenance and modulation of neuronal [2][3][4][2,3,4] and muscular (both cardiac and skeletal) excitability [5][6][7][5,6,7], regulation of calcium signaling cascades (reviewed by Reference [8]), control of cell volume [9][10][9,10], immune response [11], hormonal secretion [12], and others.
The Kv channel α-subunit belongs to the six transmembrane (6-TM) family of ion channels (Figure 1a,b) in which the voltage-sensing domain (VSD) formed by transmembrane segments S1–S4 controls pore opening via the S4–S5 intracellular loop that is connected to the pore domain (PD). The PD is formed by transmembrane segments S5, a re-entrant pore loop bearing the potassium selectivity sequence TVGYG, and S6 [13]. Depolarization of the transmembrane electric potential induces a conformational change in the VSD that leads to channel activation, opening a water-filled permeation pathway permitting K+ ions to flow down their electrochemical gradient. Upon repolarization, the VSD returns to its resting state, closing the channel gate and terminating ionic flow in a process called deactivation. Immediately after deactivation, channels can be reactivated; however, if depolarization-induced channel activation extends beyond a few milliseconds, inactivation ensues, ceasing K+ permeability. Kv channels recover from inactivation only after spending enough time at a hyperpolarized potential [14]. The molecular underpinnings of the inactivation processes have been thoroughly examined functionally and structurally, identifying various inactivation types involving distinct and complex molecular mechanisms. Voltage-gated ion channels can inactivate from pre-open closed-states (closed-state inactivation, CSI) or from the open state(s) (open-state inactivation, OSI) [15]. Inactivation can also be categorized depending on the speed of its onset upon activation. In some Kv channels, fast inactivation or N-type inactivation occurs soon after the channel activates and it is mainly due to intracellular block by the channel’s cytoplasmic N-terminus hence known as the inactivation particle [16]. This process has been directly observed by cryo-electron microscopy (cryo-EM) in a related prokaryotic K+ channel [17]. In addition to N-type inactivation, a common but relatively slower process happens after tens or hundreds of milliseconds from channel activation that is termed C-type (or slow) inactivation [18]. Even though the extent and complexity of slow inactivation remain the subjects of investigation, the permeating ions and deformations of the channel's selectivity appear to play a vital role [19]. Recent structural and functional studies support a mechanism through which the redistribution of structural water molecules accompanies the rearrangement of amino acids within the channel’s inner cavity and outer vestibule, ultimately leading to the collapse of the permeation pathway in C-type inactivation (reviewed in Reference [20]). Modulation of the inactivation process is a powerful strategy to control the cellular availability of Kv channel-mediated currents; thus, both N- and C-type inactivation are responsive to the cellular redox environment [21]. For instance, structural motifs within the Kv channel’s N-terminus/inactivation particle serve as sensors of the cytoplasmic redox potential [22].
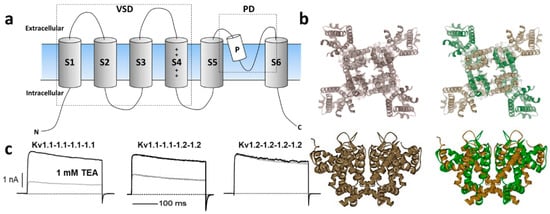
Figure 1. (a) Schematic illustration of Kv channel membrane topology depicting the 6 transmembrane segments that compose the voltage-sensing domain (VSD: S1–S4) and the pore domain (PD) formed by S5, the P-loop and S6. (b) Top and side views of representative homomeric and heteromeric Kv1 channels based on the crystal structure of Kv1.2 [(Protein Data Bank number, PDB: 2A79)] [13]. (c) Current trances of homomeric Kv1.1 (left) and Kv1.2 (right) channels and their heteromeric combination (middle) reveal distinct sensitivity to the classical pharmacological tool tetraethylammonium (TEA) [23][24][23,24].
K+ channels are the most diverse family of ion channels in excitable and non-excitable tissues, encompassing 40 Kv members implicated in many neurological, cardiac, and autoimmune disorders, which position them as important therapeutic targets [25]. They are allocated into 12 subfamilies: Kv1 (Shaker); Kv2 (Shab); Kv3 (Shaw); Kv4 (Shal); Kv7 (KvLQT); Kv10 (HERG); Kv11 (EAG); Kv12 (ELK); and modulatory/silent (KvS) Kv5, Kv6, Kv8, and Kv9 (https://doi.org/10.2218/gtopdb/F81/2019.4). The Shaker-related Kv1 family is comprised of eight members (Kv1.1–Kv1.8) encoded by the corresponding KCNA1–KCNA8 genes. Several Kv1 channels have been identified and functionally characterized within their native tissues with the help of selective blockers (reviewed in References [2][26][27][2,26,27]). For instance, Kv1 complexes were purified from the mammalian brain for the first time using dendrotoxins (DTX) from snake venom. These studies indicated that the functional Kv1 channel is a large (Mr ~400 kDa) sialoglycoprotein complex consisting of four pore-forming α-subunits and four cytoplasmically associated auxiliary β-proteins [28] that modulate K+ channel activation and inactivation kinetics (for a thorough review, refer to Reference [29]).
Kv1 channels are expressed in a variety of tissues as homo- or heterotetrameric complexes (Figure 1a,b) [30]. These complexes are formed in the endoplasmic reticulum [31], where monomers are randomly recruited, assembled, and inserted in the plasma membrane [31]. The four cytoplasmic N-terminal domains interact with one another in a strictly subfamily-specific manner, thus providing the molecular basis for the selective formation of heteromultimeric channels in vivo [32][33][32,33]. The predominant pathway of tetramer formation involves subunit dimerization in the so-called "dimer of dimers", thereby creating interaction sites different from those involved in the monomer–monomer association during the oligomerization process [34].
In heterologous expression systems, all potassium voltage-gated channel subfamily A gene (KCNA) transcripts encoding Kv1 α-subunits yield functional homo-tetrameric complexes with distinct biophysical and pharmacological profiles [35], (Figure 1c). While theoretically the combination of different Kv1 subunits could afford impressive functional diversity, only a subset of oligomeric combinations has been elucidated [36][37][38][39][40][36,37,38,39,40], suggesting their synthesis and/or assembly are carefully orchestrated. For example, amongst the KV1 channels, Kv1.2 is the most prevalent isoform in neuronal membranes where only a small fraction occurs as a homo-tetramer, while the majority are hetero-tetramers with other Kv1 α-subunits [36][40][36,40]. In these preparations, Kv1.1 subunits are consistently identified in oligomers together with Kv1.2.
2. Mechanisms of Kv Channel Inhibition by Marine Toxins
The diversity of Kv1 channels and their wide distribution, including their specific expression pattern in the central and peripheral nervous systems, together with their vital function in the excitability of nerve and muscle, make them strategic targets of marine toxins. These natural products are synthesized and deployed by marine organisms to aid predation and/or self-defense [41]. Many venomous organisms block Kv channel-mediated currents crippling membrane repolarization, causing hyper-excitability and ultimately engendering paralysis in pray or foe [42].
Marine toxins exploit different Kv channel traits to exert their modulatory actions. A commonly used strategy relies on direct occlusion of the narrow potassium permeation pathway from the extracellular side of the channel protein (Figure 2a,b). Toxins inhibiting ionic current via this mechanism are referred to as “pore blockers”. Many structurally and phylogenetically unrelated pore-blocking toxins of Kv channels share a dyad motif composed of a lysine (positive) and a tyrosine/phenylalanine (hydrophobic) [43][44][45][43,44,45]. The lysine residue fits snugly in the Kv channel selectivity filter, sterically occluding K+ ion flow, whilst the hydrophobic amino acid in the dyad aids docking and consolidation of the toxin binding. This dyad motif has been proposed to be the minimal core domain of the Kv channel-binding pharmacophore (Figure 2b) [46][47][48][49][46,47,48,49].
Mutational analysis and docking calculations have demonstrated that some marine toxins do not possess the canonical functional dyad or do not seem to use one in the classic “pore blocker” fashion to prevent potassium permeation. In these toxins, a positively charged ring of amino acids participates in electrostatic interactions with the Kv1 outer vestibule. These residues provide surface recognition and anchoring, and concurrently, a network of hydrogen bonds and hydrophobic interactions consolidate “capping” of the channel vestibule, with the peptide toxin acting as a lid over the Kv channel pore (Figure 2b) [50].
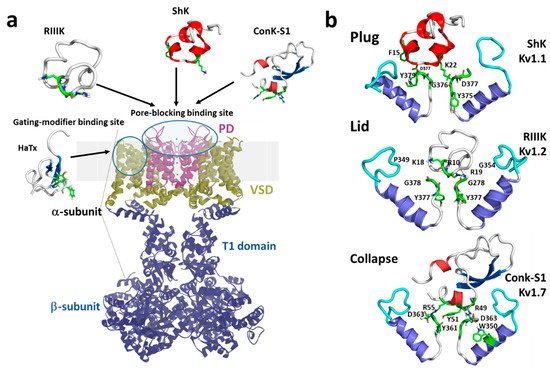
Figure 2. (a) Schematic presentation of a side view KV1 channel showing the site of interaction with representative pore-blocking peptide toxins from Cone snail (κM-RIIIK, [51] and ConK-S1, PDB: 2CA7, [52]) and sea anemone ShK (PDB: 1ROO, [53]) and gating modifier toxin from spider (HaTx; PDB: 1D1H, [54]). (b) The modes of pore-blocking (plug, lid, or collapse) are illustrated by marine peptide blockers as revealed by the docking models. The outer turret regions (residues 348–359 for Kv1.1, 350–359 for Kv1.2, and 334–343 for Kv1.7) are in cyan, and the inner turret regions (residues 377–386 for Kv1.1, 377–386 for Kv1.2, and 462–469 for Kv1.7) are indicated in green. Only two subunits of the Kv1 channels are shown, for simplicity. Docking was performed using the Haddock web server [55][56][55,56] and the docking model image was generated using Pymol (The PyMOL Molecular Graphics System, [57]).
A distinct mechanism from pore block is achieved by interacting with the gating machinery of Kv channels. Toxins acting in this fashion are known as “gating modifiers”. The VSD in Kv channels controls pore opening; hence, toxins binding to the extracellularly exposed linker between transmembrane segments S3 and S4, also known as paddle motif, within the VSD, inhibit channel function by increasing the energy required to open the channel’s gate observed as a shift in the voltage dependence of activation to more depolarized potentials. Alternatively, some toxins destabilize the Kv channel open state reflected as enhanced entry into a nonconductive inactivated state at potentials where Kv activity would normally be favored [58]. This modulatory mechanism was first shown for Hanatoxin, a gating modifier peptide component of the Chilean rose-hair tarantula venom that inhibits Kv2.1 channels (Figure 2a) [59].
A recently proposed inhibitory mechanism appears as a hybrid strategy between pore blockade and gating modification. A toxin sitting in the channel’s outer vestibule blocks the extracellular side of the permeation pathway and modifies the permeation of water molecules into proteinaceous peripheral cavities in the channel. This creates asymmetries in the distribution of water molecules around the selectivity filter, triggering a local collapse of the channel pore akin to Kv C-type inactivation (Figure 2b) [60].