The entryarticle tries to summarize our current understanding of the role of aminopeptidases in the control of blood pressure, through their effects on kidney function. Their possible role as biomarkers on acute or chronic kidney injury is also analyzed.
- renin angiotensin system
- Blood pressure
- renal biomarkers
1. Aminopeptidases in the Renin–Angiotensin System
The renin–angiotensin system (RAS) plays an essential role in blood pressure (BP) control, via vascular, renal, brain, and other mechanisms. Abnormalities in RAS activity may lead to the development of arterial hypertension and other cardiovascular and renal diseases. Blockade of this system is an effective therapeutic measure against numerous diseases, and RAS compounds have been found in the kidney, brain, and other tissues. Over recent years, our knowledge of the components of the RAS has increased, including numerous angiotensin peptides with diverse biological activities mediated by different receptor subtypes [1][2].
The enzymatic cascade of the RAS is depicted in Figure 1. It is initiated by angiotensinogen, an alfa 2 globulin of hepatic origin, which generates angiotensin I (AngI) through the enzymatic action of renin on the extreme amino-terminus. The decapeptide AngI is a substrate for angiotensin-converting enzyme (ACE), which splits the dipeptide His-Leu from the extreme carboxy-terminus to generate the octapeptide AngII [1], the major effector peptide of the RAS, which bind to two major receptors, AT1 and AT2, that generally oppose each other.
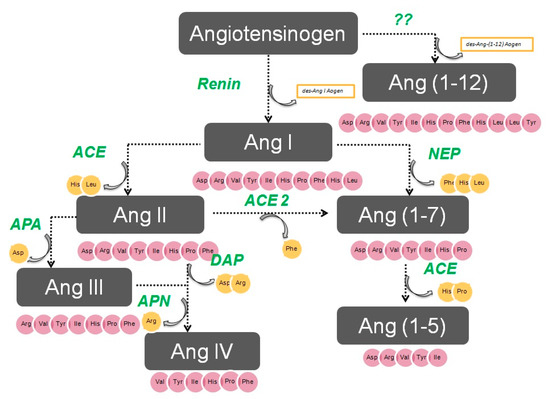
Figure 1. The renin–angiotensin system.
Action of glutamyl aminopeptidase (APA) on AngII removes the Asp residue N-terminus to generate the heptapeptide AngIII. AngIII can also be generated by an AngII-independent pathway via the nonapeptide [des-Asp1]AngI, produced from AngI by AspAP, which is converted to AngIII via ACE. APA also participates in the formation of Ang(1-7), which can also be transformed into Ang(2-7) through cleaving of the Asp-Arg bond.
In the extreme N-terminus, membrane alanyl aminopeptidase N (APN) removes Arg to give hexapeptide angiotensin IV (AngIV). AngIV is also transformed into Ang(3-7) by carboxypeptidase P (Carb-P) and propyl oligopeptidase through scission of the Pro-Phe carboxy-terminus.
ACE2 can also transform AngII into Ang(1-7) through hydrolysis of Phe by Carb-P or through scission of the dipeptide Phe-His from Ang(1-9) [1][2][3][4] .
Aminopeptidase B (APB), also known as arginine aminopeptidase (Arg-AP), cleaves basic amino acids at the N-terminus. It participates in the conversion of AngIII to AngIV.
Although not part of the RAS, both neuropeptides oxytocin and vasopressin are cleaved at the N-terminus of cysteine next to tyrosine by cystinyl aminopeptidase (CAP), a rat homolog of insulin-regulated AP (IRAP) [5].
All these enzymes are globally called “angiotensinases’’ because they are responsible for the generation of systemic and local peptides (angiotensins) related to the regulation of BP and the excretion of sodium and water. These enzymes determine the proportions of bioactive compounds.
AngI is biologically inactive, but AngII and AngIII act as agonists for AT1 and AT2 receptors, thereby mediating pressor and dipsogenic effects [6][7]. AngIV has a low affinity for AT1 and AT2 receptors but high affinity and specificity for the AT4 receptor subtype. Interaction with the AT1 receptor subtype reduces the pressor effect of AngIV. A counterregulatory system to the AngII-AngIII/AT1 receptor system is composed of ACE2 and Ang(1-7), which activate the Mas receptor [6][7][8]. This system induces vasodilatory, antifibrotic, antihypertrophic, and antiproliferative effects.
2. Aminopeptidases in Arterial Hypertension
A list of the most common aminopeptidases with their enzyme commission (EC) numbers and their main abbreviations is shown in Table 1. As was introduced in the previous paragraph, aminopeptidases (Aps) generate the active compounds of the RAS and play an essential role in BP control and sodium handling [9]. APs can also degrade certain peptidergic hormones or neuropeptides such as vasopressin, cholecystokinin, and enkephalins [10]. For this reason, APs have been analyzed in plasma, kidney, and other tissues related to BP control in several rat models of hypertension.
| Enzyme | EC Number | Abbreviations |
|---|---|---|
| Leucyl AP | 3.4.11.1 | LAP |
| Membrane alanyl AP | 3.4.11.2 | APN, AlaAP |
| Cystinyl AP | 3.4.11.3 | CysAP, CAP |
| Prolyl AP | 3.4.11.5 | PIP |
| Aminopeptidase B | 3.4.11.6 | APB, ArgAP |
| Glutamyl AP | 3.4.11.7 | APA, GluAP, EAP |
| Aminopeptidase P | 3.4.11.9 | APP |
| Cytosol alanyl AP | 3.4.11.14 | AAP, AlaAP |
| Methionyl AP | 3.4.11.18 | eMetAP |
| Aspartyl AP | 3.4.11.21 | AspAP, DNPEP |
| Arginyl AP | 3.4.22.16 | iRAP, APR |
Thus, reduced renal membrane-bound APA, iRAP, and APN activities were observed in a reduced renal mass saline model, and reduced APA activity was detected in a two-kidney one-clip Goldblatt hypertension model [11]. In the low renal mass model, a positive correlation was found in both soluble CAP and APN activities between the neurohypophysis and the adrenal gland, but this was not observed in the normotensive rats [12].
The relationship between APs and arterial hypertension has been explored with greater precision. For instance, it has been proposed that endoplasmic reticulum AP 1 (ERAP1) and ERAP2 regulate BP by inactivation of AngII, and these two APs, which also hydrolyze amino acids from the N-terminus of various human antigens and peptide hormones, are widely expressed in human tissues, including heart, endothelial cells, and kidney [13][14][15]. In vitro, ERAP1 transforms AngII into AngIII and AngIV [14], while ERAP2 converts AngIII to AngIV [15]. ERAP 1 was initially identified as a placental leucine AP that degrades AngII and III and transforms kallidin into bradykinin in vitro, thus has a role in BP regulation [14].
In this sense, several interesting papers have been published. In an in vivo study, an increase in circulating levels of ERAP1 was found to reduce BP and AngII levels [16]. In a genetic study, an association was detected between variants of the gene encoding Arg528 and the development of essential hypertension in a Japanese population [17]. In an investigation of 45 genetic variants of ERAP1 and ERAP2 in 17,255 Caucasian females from the Women’s Genome Health Study, ERAP1 was found to be related to increased BP [18]. The ERAP1 genotype also appears to be involved in the reduction of left ventricular mass produced by some antihypertensive treatments [19]. Thus, all these data indicate a possible role for ERAP1 in BP regulation and ventricular remodeling. Finally, other genetic studies have associated variants of ERAP1 and ERAP2 genes with preeclampsia [20], hemolytic uremia [21], and hypertension [17].
Aminopeptidases after Antihypertensive Therapy
Various systemic antihypertensive treatments can alter the activities of brain and systemic APs associated with effects on BP. Thus, in a rat study by Banegas et al. [22], unilateral brain lesions in the nigrostriatal system produced simultaneous and paralleled changes in BP and in brain and plasma AP activities. Later, the same group [23] examined the participation of APs in the metabolism of some angiotensins, vasopressin, cholecystokinin, and enkephalins in the plasma and hypothalamus of spontaneous hypertensive (SHR) rats under normal conditions and after beta-blocker treatment with propranolol. In this rat strain, AP activity in response to propranolol administration differed between plasma and hypothalamus, thus suggesting an interaction between APs and the autonomic nervous system. Moreover, these authors also reported that treatment with ACE inhibitors captopril, propranolol, or the nitric oxide synthesis inhibitor L-NAME, induced a marked modification of brain patterns of neuropeptidase activity in SHR rats [24].
The BP-lowering effect of a diet enriched in extra virgin olive oil in rats was analyzed by Villarejo et al. [25] who observed that rats receiving this diet showed augmented APN and AspAP activity in the renal cortex, suggesting a greater degradation of AngIII and AngIV and an increased generation of the antihypertensive Ang(2-10). Hence, the glomerular formation of Ang(2-10) might compensate the well-known pressor effects of AngII on the glomerular vasculature in this model of hypertension.
Taken together, the data reported in this section indicate that BP changes induced by antihypertensive or prohypertensive drugs are associated with modifications in AP activity. However, no definitive conclusions can be drawn about the functional role of APs in the pathogenesis of hypertension.
3. Therapeutic Strategies to Treat Arterial Hypertension with Aminopeptidases
The vast majority of studies on the control of BP and treatment of hypertension have addressed the blockade of AngII or its receptors, and there has been less research on the regulation of other angiotensin peptides. Thus, blockade of the brain RAS has been found to simultaneously decrease sympathetic tone, vasopressin release, and baroreflex activity, thereby reducing cardiac output and peripheral resistance [26].
An action on central or peripheral APs represents a new approach to the treatment of hypertension. Thus, new antihypertensive treatments have been developed based on potent orally-active inhibitors of APA or activators of aspartyl-aminopeptidase (DNPEP), since brain aspartyl aminopeptidase exerts BP-lowering effects by transforming AngI into angiotensin 2-10. Currently, the search for new antihypertensive compounds that affect the RAS multi-enzyme cascade is an important line of research.
3.1. Inhibition of APA
The icv administration of EC33, a specific APA inhibitor, prevented the BP increase produced by the icv administration of AngII in SHR animals, indicating that the central response to AngII requires its transformation into AngIII by APA. A marked BP decrease has also been observed in conscious SHR and DOCA-salt hypertensive rats after the icv infusion of EC33 [27][28]. In contrast, the peripheral iv infusion of EC33 did not reduce BP, indicating that EC33 does not cross the blood–brain barrier and/or is inactive in systemic circulation.
The BP of SHR rats increased after the central icv administration of APA [29], probably due to an increased endogenous generation of AngIII, whereas APA blockade with an antiserum attenuated the pressor response to AngII by around 60% [29]. It is interesting to note that a selective APA inhibitor (RB150) with antihypertensive properties can be given either intravenously [28] or orally [30] because it can cross the blood–brain barrier.
The peripheral activity of the AT1 receptor depends on the transformation of AngII into AngIII by APA [31]. Thus, antihypertensive effects were observed in SHR rats after the systemic administration of recombinant APA [32] at a dose that was one-tenth of the usual candesartan dose [33]; the joint i.v. administration of APA and APN attenuated the pressor effect of AngII in normal rats and treatment with APA reduced the BP of SHR rats to normal levels [34].
Considered together, these data clearly demonstrate that APA reduces BP, while abnormalities in APA activity promote hypertension, as supported by the lower renal APA activity in SHR versus WKY rats [35]. The administration of APA has therefore been proposed for the treatment of acute heart failure, acute hypertensive crisis, preeclampsia, and hypertensive encephalopathy, among other hypertensive emergencies [36][37].
3.2. APN Blockade in the Treatment of Hypertension
The administration of PC18, an inhibitor of APN, generates a pressor response through the accumulation of endogenous AngIII, which is mediated via the AT1 receptor. In this way, pretreatment with the AT1 blocker losartan can suppress the pressor response, while the AT2 antagonist PD123319 is unable to prevent the BP increase [38]. The enhanced proximal tubular sodium reabsorption of SHR rats is prevented by the intrarenal infusion of PC18 [39]. This finding indicates that the blockade of AngIII degradation achieved by APN inhibition improved sodium excretion in the proximal tubule of these rats when it was administered in the renal interstitium [40]. Hence, the transformation of AngII into AngIII is required for this natriuretic response, which is not affected by the AT1 blocker candesartan. Research on the usefulness of APN inhibitors to treat hypertensive patients is at an early stage, and further studies are required.
4. APs as Urinary Biomarkers of Renal Injury
Serum creatinine and blood urea nitrogen (BUN) are widely used markers of renal disease, but their sensitivity and specificity are limited, and they are not useful in distinguishing the stages of acute kidney injury (AKI) [41]. They lack sensitivity because they increase only when the renal lesion is evident. Thus, serum creatinine levels rise gradually, and the kidneys have already lost half of their functionality by the time normal levels are doubled [42][43]. Besides, normal levels of these markers can be affected by protein-rich diets, intestinal bleeding, muscle disease, and dehydration, generating false positives in the diagnosis of renal disease. There is a need for biomarkers to allow an earlier diagnosis of AKI, a better prediction of renal disease severity, and an improved assessment of adverse effects in drug development [41].
Various urinary biomarkers for the early detection of AKI have emerged over recent years [42][43], including tubular enzymes that are increased in urine after damage to the tubular epithelium [43], which can precede or even trigger renal dysfunction. Major advantages of urinary markers include the non-invasiveness of the sampling and their usefulness to elucidate the size and localization of tubular cell lesions and to detect the presence of necrosis or other alterations that evoke renal dysfunction [44]. The measurement of urinary enzymes and other urinary biomarkers may therefore be a valuable tool to obtain an early diagnosis during initial stages of renal disease and to follow its progression or regression, facilitating prediction of the prognosis. Urinary APs are considered promising and useful biomarkers of renal disease of different pathophysiological origin, and an automated photometric assay has been developed for their measurement [45].
APA, APN, and CAP are present in the brush border membrane of renal tubular cells [46], have molecular weights above 140 kDa, and are highly organ-specific. These conditions ensure the tubular origin of these urinary enzymes, which cannot pass easily into the urine through the glomerular barrier (Figure 2).
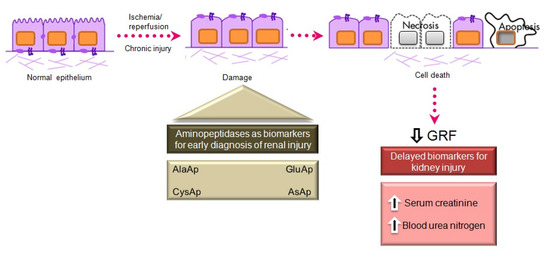
Figure 2. Main aminopeptidases studied as biomarkers of acute and chronic kidney injury.
APs participate in AngII metabolism, forming part of the renal renin–angiotensin–aldosterone system (RAAS) [47], which is elevated in renal diseases. One of these enzymes, alanine aminopeptidase (APN), is a brush border enzyme that was proposed in the early 1970s as a urinary marker of renal disease [48]. Later, Marchewka et al. [49] demonstrated that the measurement of APN and its isoforms was of diagnostic relevance in nephrolithiasis. They also observed significantly higher APN excretion in patients with glomerulonephritis than in controls and a positive correlation between urinary protein concentrations and APN activity [50]. Their findings supported the association between proteinuria and elevated activity of renal tubular brush border enzymes reported in other studies of chronic glomerulonephritis [51]. As an explanation of this association, these authors [50] proposed that the protein present in the ultrafiltrate produces a release of APN from the external membrane of renal tubule microvilli. Because of its external localization, its release into the urine can be caused by a weak destructive action and is not necessarily linked to disruption of the integrity of kidney tubule cells [52].
Jung et al. [53] reported that the urinary excretion of APN, alkaline phosphatase, γ-glutamyltransferase, and N-acetyl-β-D-glucosaminidase is age-dependent in humans. These enzymes were determined in random morning urine samples from 442 individuals aged from 5 days to 58 years, and their creatinine-normalized activity significantly decreased with increasing age. APN activity has also been proposed as a biomarker of the nephrotoxicity induced by vancomycin [52] or amphotericin B [54] in experimental models and in several human diseases, such as glomerulopathy [55], IgA nephropathy [56], and diabetes [57]. However, conflicting results have been published on its usefulness as a biomarker of renal function in kidney transplantation patients [58][59].
Acute Kidney Injury
AKI is defined by an abrupt increase in serum creatinine during the 48 h period after an insult responsible for functional or structural changes in the kidney, and it is mainly caused by the acute apoptosis or necrosis of renal tubular cells [60]. The early detection of AKI is a current preclinical and clinical research priority. The administration of nephrotoxic drugs that alter tubular function is the most common experimental model to relate the excretion of different biomarkers to renal dysfunction. Cisplatin is an antineoplastic drug with a potent nephrotoxic effect, mainly due to its action on the proximal tubule, causing AKI in experimental animal models and patients [61][62]. Its administration induces tubular and glomerular disturbances that lead to the development of interstitial fibrosis [63].
Our group investigated whether urinary activities of APN, APA, and CAP could serve as biomarkers of renal dysfunction by using a rat model of cisplatin-induced nephrotoxicity [64]. Their activity was determined in urine samples collected at 24, 48, and 72 h after cisplatin injection. Their urinary activity at 24 h post-injection was correlated with two renal function indicators (plasma creatinine and creatinine clearance) and their activity at two weeks post-injection was correlated with two indicators of structural damage (renal hypertrophy and interstitial fibrosis). A comparative analysis was also performed with other proposed urinary biomarkers of kidney injury, i.e., albumin, proteinuria, NAG, and NGAL. The area under the curve (AUC) was calculated to quantify the sensitivity and specificity of each marker to distinguish cisplatin-treated from control rats at 24 h post-injection (Figure 3). APN, APA, CAP, and DNPEP had an AUC value greater than 0.5, indicating that these enzymes are early biomarkers of kidney injury in cisplatin-treated rats. The greatest specificity and sensitivity values were observed for APN, which was the best variable to discriminate between treated and untreated animals, whereas an AUC value below 0.5 was observed for NAG and NGAL at this time point. A significant correlation was also found between AP activities at day 1 post-injection and functional and structural variables at day 14. It can therefore be concluded that the urinary activities of these APs may serve as early and predictive biomarkers of cisplatin-induced renal dysfunction. The measurement of urinary AP activities can be useful in preclinical research for the early detection and follow-up of renal damage and for the evaluation of drug nephrotoxicity, and it may offer a useful prognostic and diagnostic tool for renal diseases in the clinical setting.
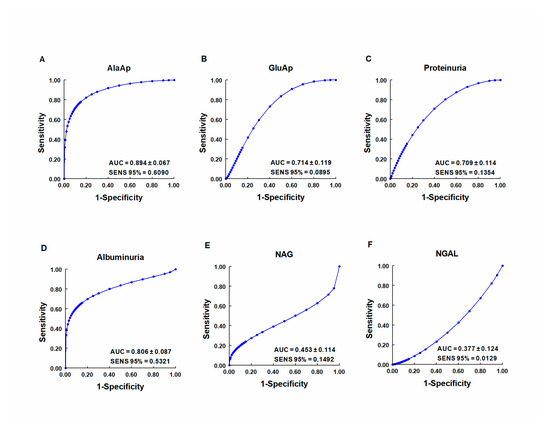
Figure 3. ROC curves showing specificity and sensitivity for APN (AlaAp) (A), APA (GluAp) (B), proteinuria (C), albuminuria (D), N-acetyl-β-D-glucosaminidase (NAG) (E), and neutrophil gelatinase-associated lipocalin (NGAL) (F) to differentiate cisplatin-treated rats from control rats 24 h after injection of saline, 3.5 or 7 mg/kg of cisplatin (n = 8 each group). All cisplatin-treated rats displayed tubular dysplasia and interstitial fibrosis 14 days after injection. Urinary markers were expressed in daily total activity or excretion per 100 g of body weight. AUC = area under the curve. SENS 95 % = calculated sensitivity at 95 % of specificity.
References
- Johnston, C.I. Biochemistry and pharmacology of the renin-angiotensin system. Drugs 1990, 39, 21–31, doi:10.2165/00003495-199000391-00005.
- Reudelhuber, T.L. The renin-angiotensin system: Peptides and enzymes beyond angiotensin II. Curr. Opin. Nephrol. Hypertens. 2005, 14, 155–159, doi:10.1097/00041552-200503000-00011.
- Ferrario, C.M.; Chappell, M.C. Novel Angiotensin peptides. Cell. Mol. Life. Sci. 2004, 61, 2720–2727.
- Vauquelin, G.; Michotte Smolders, Y.I.; Sarre, S.; Ebinger, G.; Dupont, A.; Vanderheyden, P. Cellular targets for angiotensin II fragments: Pharmacological and molecular evidence. J. Renin Angiotensin Aldosterone Syst. 2002, 3, 195–204.
- Enzyme Nomenclature. Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes by the Reactions they Catalyse. Available online: https://www.qmul.ac.uk/sbcs/iubmb/enzyme/ (accessed on 18 June 2020).
- De Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472.
- Dinh, D.T.; Frauman, A.G.; Johnston, C.I.; Fabiani, M.E. Angiotensin receptors: Distribution, signalling and function. Clin. Sci. 2001, 100, 481–492.
- Chappell, M.C. Nonclassical renin-angiotensin system and renal function. Compr. Physiol. 2012, 2, 2733–2735.
- Prieto, I.; Villarejo, A.B.; Segarra, A.B.; Banegas, I.; Wangensteen, R.; Martinez-Cañamero, M.; de Gasparo, M.; Vives, F.; Ramírez-Sánchez, M. Brain, heart and kidney correlate for the control of blood pressure and water balance: Role of angiotensinases. Neuroendocrinology 2014, 100, 198–208.
- Ramírez, M.; Prieto, I.; Alba, F.; Vives, F.; Banegas, I.; de Gasparo, M. Role of central and peripheral aminopeptidase activities in the control of blood pressure: A working hypothesis. Heart Fail. Rev. 2008, 13, 339–353.
- Ramírez, M.; Prieto, I.; Martinez, J.M.; Vargas, F.; Alba, F. Renal aminopeptidase activities in animal models of hypertension. Regul. Pept. 1997, 72, 155–159.
- Prieto, I.; Martinez, A.; Martinez, J.M.; Ramírez, M.J.; Vargas, F.; Alba, F.; Ramírez, M. Activities of aminopeptidases in a rat saline model of volume hypertension. Horm. Metab. Res. 1998, 30, 246–248.
- Tsujimoto, M.; Goto, Y.; Maruyama,M.; Hattori, A. Biochemical and enzymatic properties of the m1 family of aminopeptidases involved in the regulation of blood pressure. Heart Fail. Rev. 2008, 13, 285–291.
- Hattori, A.; Kitatani, K.; Matsumoto, H.; Miyazawa, S.; Rogi, T.; Tsuruoka, N.; Mizutani, S.; Natori, Y.; Tsujimoto, M. Characterization of recombinant human adipocyte-derived leucine aminopeptidase expressed in Chinese hamster ovary cells. J. Biochem. 2000, 128, 755–762.
- Tanioka, T.; Hattori, A.; Masuda, S.; Nomura, Y.; Nakayama, H.; Mizutani, S.; Tsujimoto, M. Human leukocytederived arginine aminopeptidase. The third member of the oxytocinase subfamily of aminopeptidases. J. Biol. Chem. 2003, 278, 32275–32283.
- Hisatsune, C.; Ebisui, E.; Usui, M.; Ogawa, N.; Suzuki , A.; Mataga , N.; Hiromi Takahashi-Iwanaga , H.; Katsuhiko Mikoshiba, K. ERp44 exerts redox dependent control of blood pressure at the ER. Mol. Cell 2015, 58, 1015–1027.
- Yamamoto, N.; Nakayama, J.; Yamakawa-Kobayashi, K.; Hamaguchi, H.; Miyazaki, R.; Arinami, T. Identification of 33 polymorphisms in the adipocyte-derived leucine aminopeptidase (ALAP) gene and possible association with hypertension. Hum. Mutat. 2002, 19, 251–257.
- Robert, Y.; Zee, L.; Rivera, A.; Inostroza, Y.;Ridker, P.M.; Daniel, I.; Chasman, D.I.; Romero, J.R. Gene variation of endoplasmic reticulum aminopeptidases 1 and 2, and risk of blood pressure progression and incident hypertension among 17,255 initially healthy women. Intern. J. Genom. 2018, 2018, 2308585.
- Hallberg, P.; Lind, L.; Michaelsson, K.; Kurland, L.; Kahan, T.; Malmqvist, K.; Ohman, K.P.; Nystrom, F.; Liljedahl, U.; Syvanen, A.C.; et al. Adipocyte-derived leucine aminopeptidase genotype and response to antihypertensive therapy. BMC Cardiovasc. Disord. 2003, 3, 11.
- Johnson, M.P.; Roten, L.T.; Dyer, T.D.; East, C.E.; Forsmo, S.; Blangero, J.; Brennecke, S.P.; Austgulen, R.; Moses, E.K. The ERAP2 gene is associated with preeclampsia in Australian and Norwegian populations. Hum. Genet. 2009, 126, 655–666.
- Taranta, A.; Gianviti, A.; Palma, A.; De Luca, V.; Mannucci, L.; Procaccino, M.A.; Ghiggeri, G.M.; Caridi, G.; Fruci, D.; Ferracuti, S.; et al.Genetic risk factors in typical haemolytic uraemic syndrome. Nephrol. Dial. Transplant. 2009, 24, 1851–1857.
- Banegas, I.; Ramírez, M.; Vives, F.; Alba, F.; Segarra, A.B.; Duran, R.; De Gasparo, M.; Prieto, I. Aminopeptidase activity in the nigrostriatal system and prefrontal cortex of rats with experimental hemiparkinsonism. Horm. Metab. Res. 2005, 37, 53–55.
- Prieto, I.; Segarra, A.B.; de Gasparo, M.; Martínez-Cañamero, M.; Ramírez-Sánchez, M. Divergent profile between hypothalamic and plasmatic aminopeptidase activities in WKY and SHR. Influence of beta-adrenergic blockade. Life Sci. 2018, 192, 9–17.
- Prieto, I.; Segarra, A.B.; Villarejo, A.B.; de Gasparo, M.; Martínez-Cañamero, M.M.; Ramírez-Sánchez, M. Neuropeptidase activity in the frontal cortex of Wistar-Kyoto and spontaneously hypertensive rats treated with vasoactive drugs: A bilateral study. J. Hypertens. 2019, 37, 612–628.
- Villarejo, A.B.; Ramírez-Sánchez, M.; Segarra, A.B.; Martínez-Cañamero, M.; Prieto, I. Influence of extra virgin olive oil on blood pressure and kidney angiotensinase activities in spontaneously hypertensive rats. Planta Med. 2015, 81, 664–669.
- Marc, Y.; Llorens-Cortes, C. The role of the brain renin-angiotensin system in hypertension: Implications for new treatment. Prog. Neurobiol. 2011, 95, 89–103.
- Morton, J.J.; Casals-Stenzel, J.; Lever, A.F. Inhibitors of the renin-angiotensin system in experimental hypertension, with a note on the measurement of angiotensin I, II and III during infusion of converting-enzyme inhibitor. Br. J. Clin. Pharmacol. 1979, 2, 233S–241S.
- Fournie-Zaluski, M.C.; Fassot, C.; Valentin, B.; Djordjijevic, D.; Reaux-Le Goazigo, A.; Corvol, P.; Roques, B.P.; Llorens-Cortes, C. Brain renin-angiotensin system blockade by systemically active aminopeptidase A inhibitors: A potential treatment of salt-dependent hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 7775–7780.
- Song, L.; Wilk, S.; Healy, D.P. Aminopeptidase A antiserum inhibits intracerebroventricular angiotensin IIinduced dipsogenic and pressor responses. Brain Res. 1997, 744, 1–6.
- Bodineau, L.; Frugière, A.; Marc, Y.; eInguimbert, N.; Fassot, C.; Balavoine, F.; Roques, B.; Llorens-Cortes, C. Orally active aminopeptidase A inhibitors reduce blood pressure: A new strategy for treating hypertension. Hypertension 2008, 51, 1318–1325.
- Wright, J.W.; Harding, J.W. Brain renin-angiotensin-A new look at an old system. Prog. Neurobiol. 2011, 95, 49–67.
- Goto, Y.; Hattori, A.; Ishii, Y.; Tsujimoto, M. Reduced activity of the hypertension-associated Lys528Arg mutant of human adipocyte-derived leucine aminopeptidase (ALAP)/ ER-aminopeptidase-1. FEBS Lett. 2006, 580, 1833–1838.
- Ishii, M.; Hattori, A.; Numaguchi, Y.; Tsujimoto, M.; Ishiura, S.; Kobayashi, H.; Murohara, T.; Wrght, J.W.; Mizutani, S. The effect of recombinant aminopeptidase A on hypertension in spontaneously hypertensive rats: Its effect in comparison with candesartan. Horm. Metab. Res. 2008, 40, 887–891.
- Mizutani, S.; Okano, K.; Hasegawa, E. Human placental leucine aminopeptidase (P-LAP) as a hypotensive agent. Experientia 1982, 38, 821–822.
- Nakashima, Y.; Ohno, Y.; Itakura, A.; Takeuchi, M.; Murata, Y.; Kuno, N.; Mizutani, S. Possible involvement of aminopeptidase A in hypertension in spontaneously hypertensive rats (SHRs) and change of refractoriness in response to angiotensin II in pregnant SHRs. J. Hypertens. 2002, 20, 2233–2238.
- Mizutani, S.; Wright, J.; Kobayashi, H. A new approach regarding the treatment of preeclampsia and preterm labor. Life Sci. 2011, 88, 17–23.
- Kobayashi, H.; Mizutani, S.; Wright, J.W. Placental leucine aminopeptidase- and aminopeptidase A-deficient mice offer insight concerning the mechanisms underlying preterm labor and preeclampsia. J. Biomed. Biotechnol. 2011, 2011, 286947.
- Carey, R.M.; Padia, S.H. Role of angiotensin at2 receptors in natriuresis: Intrarenal mechanisms and therapeutic potential. Clin. Exp. Pharmacol. Physiol. 2013, 40, 527–534, doi:10.1111/1440-1681.12059.
- Padia, S.H.; Howell, N.L.; Kemp, B.A.; Fournie-Zaluski, M.C.; Roques, B.P.; Carey, R.M. Intrarenal aminopeptidase N inhibition restores defective angiontesin II type 2-mediated natriuresis in spontaneously hypertensive rats. Hypertension 2010, 55, 474–480.
- Padia, S.H.; Kemp, B.A.; Howell, N.L.; Gildea, J.J.; Keller, S.R.; Carey, R.M. Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension 2009, 53, 338–343.
- Vaidya, V.S.; Ferguson, M.A.; Bonventre, J.V. Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 463–493.
- Bonventre, J.V.; Vaidya, V.S.; Schmouder, R.; Feig, P.; Dieterle, F. Nextgeneration biomarkers for detecting kidney toxicity. Nat. Biotechnol. 2010, 28, 436–440.
- Devarajan, P. Emerging biomarkers of acute kidney injury. Contrib. Nephrol. 2007, 156, 203–212.
- Lisowska-Myjak, B. Serum and urinary biomarker of acute kidney injury. Blood. Purif. 2010, 29, 357–365.
- Holdt, B.; Peters, E.; Nagel, H.R.; Steiner, M. An automated assay of urinary alanine aminopeptidase activity. Clin. Chem. Lab. Med. 2008, 46, 537–540.
- Song, L.; Ye, M.; Troyanovskaya, M.; Wilk, E.; Wilk, S.; Healy, D.P. Rat kidney glutamyl aminopeptidase (aminopeptidase A): Molecular identity and cellular localization. Am. J. Physiol. 1994, 267, F546–F557.
- Segarra, A.B., Ramírez, M.; Banegas, I.; Hermoso, F.; Vargas, F.; Vives, F.; Alba, F.; de Gasparo, M.; Prieto, I. Influence of thyroid disorders on kidney angiotensinase activity. Horm. Metab. Res. 2006, 38, 48–52.
- Peters, J.E.; Mampel, E.; Schneider, I.; Burchardt, U.; Fukala, E.; Ahrens, I.; Haschen, R.J. Alanine aminopeptidase in urine in renal diseases. Clin. Chim. Acta 1972, 37, 213–224.
- Marchewka, Z.; Długosz, A.; Kúzniar, J. Diagnostic application of AAP isoenzyme separation. Int. Urol. Nephrol. 1999, 31, 409–416.
- Marchewka, Z.; Kúzniar, J., Długosz, A. Enzymuria and β2-Mikroglobulinuria in the assessment of the influence of proteinuria on the progression of glomerulopathies. Int. Urol. Nephrol. 2001, 33, 673–676.
- Idasiak-Piechocka, I.; Krzymánski, M. The role of tubulointerstitial changes in progression of chronic glomerulonephritis (GN). Przegl. Lek. 1996, 53, 443–453.
- Naghibi, B.; Ghafghazi, T.; Hajhashemi, V.; Talebi, A. Vancomycin-induced nephrotoxicity in rats: Is enzyme elevation a consistent finding in tubular injury? J. Nephrol. 2007, 20, 482–488.
- Jung, K.; Hempel, A.; Grutzmann, K.D.; Hempel, R.D.; Schreiber, G. Age-dependent excretion of alanine aminopeptydase, alkaline phosphatase, γ -glutamyltransferase and N-acetyl-β-D-glucosaaminidase in human urine. Enzyme 1990, 43, 10–16.
- Inselmann, G.; Balaschke, M.; Heidemann, H.T. Enzymuria following amphotericin B application in the rat. Mycoses 2003, 46, 169–173.
- Mitic, B.; Lazarevic, G.; Vlahovic, P.; Rajic, M.; Stefanovic, V. Diagnostic value of the aminopeptidase N, N-acetyl-beta-D-glucosaminidase and dipeptidylpeptidase IV in evaluating tubular dysfunction in patients with glomerulopathies. Ren. Fail. 2008, 30, 896–903.
- Moon, P.G.; Lee, J.E.; You, S.; Kim, T.K.; Cho, J.H.; Kim, I.S.; Kwon, T.H.; Kim, C.D.; Park, S.H.; Hwang, D.; et al. Proteomic analysis of urinary exosomes from patients of early IgA nephropathy and thin basement membrane nephropathy. Proteomics 2011, 11, 2459–2475.
- Lazarevic, G.; Antic, S.; Vlahovic, P.; Djordjevic, V.; Zvezdanovic, L.; Stefanovic, V.Effects of aerobic exercise on microalbuminuria and enzymuria in type 2 diabetic patients. Ren. Fail. 2007, 29, 199–205.
- Kuzniar, J.; Marchewka, Z.; Krasnowski, R.; Boratynska, M.; Długosz, A.; Klinger, M. Enzymuria and low molecular weight protein excretion as the differentiating marker of complications in the early post kidney transplantation period. Int. Urol. Nephrol. 2006, 38, 753–758.
- Marchewka, Z.; Kuzniar, J.; Zynek-Litwin, M.; Falkiewicz, K.; Szymanska, B.; Roszkowska, A.; Klinger, M. Kidney graft function in long-term cyclosporine and tacrolimus treatment: Comparative study with nephrotoxicity markers. Transplant. Proc. 2009, 41, 1660–1665.
- Molitoris, B.A.; Levin, A.; Warnock, D.G.; Joannidis, M.; Mehta, R. L.; eKellum, J.A.; Ronco, C.; Shah, S. Improving outcomes from acute kidney injury. J. Am. Soc. Nephrol. 2007, 18, 1992–1994.
- Safirstein, R.; Winston, J.; Moel, D.; Dikman, S.; Guttenplan, J. Cisplatin nephrotoxicity-insights into mechanism. Int. J. Androl. 1987, 10, 325–346.
- Winston, J.A.; Safirstein, R. Reduced renal blood-flow in early cisplatin induced acute renal failure in the rat. Am. J. Physiol. 1985, 249, F490–F496.
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin nephrotoxicity: A review. Am. J. Med. Sci. 2007, 334, 115–124.
- Quesada, A.; Vargas, F.; Montoro-Molina, S.; O’Valle, F.; Rodríguez-Martínez, M.D.; Osuna, A.; Prieto, I.; Ramírez, M.; Wangensteen, F. Urinary Aminopeptidase Activities as Early and Predictive Biomarkers of Renal Dysfunction in Cisplatin-Treated Rats. PLoS ONE 2012, 7, e40402.