Rheumatoid arthritis (RA) and other autoimmune inflammatory diseases are examples of imbalances within the immune system (disrupted homeostasis) that arise from the effects of an accumulation of environmental and habitual insults over a lifetime, combined with genetic predispositions. The Ligand Epitope Antigen Presentation System (LEAPS) therapies are capable of inhibiting ongoing disease progression in animal models. Whereas DMARDs ablate or inhibit specific proinflammatory cytokines or cells and JAK inhibitors (jakinibs) inhibit the receptor activation cascade for expression of proinflammatory cytokines, the LEAPS therapeutic vaccines specifically modulate the ongoing antigen-specific, disease-driving, proinflammatory T memory cell responses. This decreases disease presentation and changes the cytokine conversation to decrease the expression of inflammatory cytokines while increasing the expression of regulatory cytokines.
- immunotherapy
- inflammatory
- anti-inflammatory
- cytokines
- rheumatoid arthritis
1. From Homeostasis to Autoimmunity
2. Post-Translational Modification and Its Role in Autoimmunity
3. Role of Cytokines, Cells, and Their Interplay in Disrupting Immune Homeostasis
In the presence of systemic inflammation, potentially exacerbated by trauma, infection or other inflammatory trigger, autoimmune responses can be initiated against PTM proteins to alter the normal balance of immunity. Figure 1A represents the normal balance in immune pro-inflammatory and anti-inflammatory responses that promote immune homeostasis. The various actors include cells (T and B cells, macrophages, dendritic cells (DC), other blood cells) and pro-inflammatory and anti-inflammatory cytokines that affect the regulation of responses to self (auto)antigen.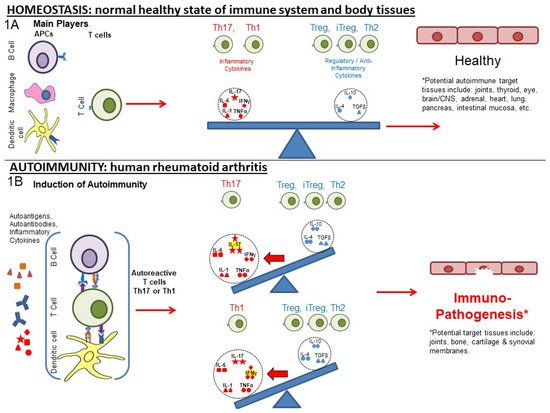
4. Current and Older Therapeutic Approaches for RA
| Type | Target | ↓/↑ Modulation | Regulated Immune Component, If Known [References] | Generic and Product name, Regulatory Status | Ref. | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Therapeutic Vaccines | Th1 | ↓ | IL-1, IL-17, IFN-γ, TNF-α [3] | IL-1 | [17] | , IL-17 | [18] | , IFN-γ, TNF-α [3,29,30] | CEL-4000 (preclinical) | [3][17][18] | [3,29,30] | |||||||||||
| ↑ | Treg (FOXP3+), IL-4, IL-10, TGF-β [3][17][18] | Treg (FOXP3+), IL-4, IL-10, TGF-β [3,29,30] | ||||||||||||||||||||
| Th17 | ↓ | TNF-α, IL-17, IL-6, MCP-1, IL-12p40 [19] | TNF-α, IL-17, IL-6, MCP-1, IL-12p40 [27] | CEL-2000 (preclinical) | [19] | [27] | ||||||||||||||||
| ↑ | IL-12p70, IL-10 [19] | IL-12p70, IL-10 [27] | ||||||||||||||||||||
| DMARDs | TNF-α | ↓ | TNF-α [20] | TNF-α [41] | Adalimumab (Humira®) | [20][21] | [41,42] | |||||||||||||||
| TNF-α | ↓ | TNF-α [22] | TNF-α [43] | Etanercept (Enbrel®) | [22] | [43] | ||||||||||||||||
| IL-1Ra | ↓ | IL-1 [23] | IL-1 [44] | Anakinra (Kineret®) | [23] | [44] | ||||||||||||||||
| IL-6R msR | ↓ | MCP-1 [21], IL-6 [24] | MCP-1 [42], IL-6 [45] | Tocilizumab (Actemra®) | [21][24] | [42,45] | ||||||||||||||||
| IL-17 | ↓ | MCP-1 [21], IL-17A [25] | MCP-1 [42], IL-17A [46] | Secukinumab (Cosentyx®) | [21][25] | [42,46] | ||||||||||||||||
| CD20 | ↓ | B cells as APCs: CD4+IFN-γ+, CD4+IL-17+ [26] | B cells as APCs: CD4+IFN-γ+, CD4+IL-17+ [47] | Rituximab (Rituxan®) | [26][27] | [47,48] | ||||||||||||||||
| Anti-CD6 | ↓ | IL-17 [28], IFN-γ [28][29], IL-6, TNF-α | IL-17 | [29] | [49], IFN-γ [49,50], IL-6, TNF-α [50] | Itolizumab (Alzumab®) | [28][29][30] | [49,50,51] | ||||||||||||||
| Agonistic Anti-CD137 | ↑ | IFN-γ [31] | IFN-γ | [32], IDO [] | [52 | 32 | ,53], IDO [53] | Utomilumab | [31][32] | [52,53] | ||||||||||||
| Anti-CTLA4 | ↓ | IL-17, IFN-γ [33] | IL-17, IFN-γ [54] | Abatacept (Orencia®) | [33][34][35] | [54,55,56] | ||||||||||||||||
| ↑ | IL-35, IFN-β [33] | IL-35, IFN-β [54] | ||||||||||||||||||||
| Anti-CD40 | ↓ | IL-6, RANKL [36], TNF-α, NF-κβ, IL-6, ICAM-1, VCAM-1, VEGF | IL-6 | [37] | , RANKL [57], TNF-α, NF-κβ, IL-6, ICAM-1, VCAM-1, VEGF [58] | Bi 655064 | [36][37] | [57,58] | ||||||||||||||
| CD24 | ↓ | TNF-α, IL-6, MCP-1(CCL2), IL-1β [38] NF-κβ | TNF-α | [39] | , IL-6, MCP-1(CCL2), IL-1β [59] NF-κβ [60] | [38][40][41] | [59,61,62] | |||||||||||||||
| Jakinibs | JAK3 > JAK1, JAK2 > TYK2 [42] | JAK3 > JAK1, JAK2 > TYK2 [63] | ↓ | Transcription: IL-2, IL-4, IL-7, IL-9, IL-15, IL-21, IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IFN-α, IFN-β, IL-10, IL-22, IFN-γ, > EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 > IL-12, IL-23, Type III IFNs [43] in vitro: IL-6 by B cells, [44] IL-2, IL-4, IL-7, IL-15, IL-21, IL-6, and IFN-γ in CD4+ T cells. IL-17 in Th17 cells polarized via IL-23. IL-21 and IL-22 in Th17 [45], IFN-α, IL-6, IFN-γ, IL-2, IL-15, IL-4, GM-CSF [43] MCP-1 [47] | : IL-2, IL-4, IL-7, IL-9, IL-15, IL-21, IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IFN-α, IFN-β, IL-10, IL-22, IFN-γ | 21] IL-17 in CD4+T cells from AS, PSA, RA, and HC [46] in vivo: IL-6 in human | , > EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 > IL-12, IL-23, Type III IFNs [64] in vitro: IL-6 by B cells, [65 | [ | ] IL-2, IL-4, IL-7, IL-15, IL-21, IL-6, and IFN-γ in CD4+ T cells. IL-17 in Th17 cells polarized via IL-23. IL-21 and IL-22 in Th17 [66], IFN-α, IL-6, IFN-γ, IL-2, IL-15, IL-4, GM-CSF [64] MCP-1 [42] IL-17 in CD4+T cells from AS, PSA, RA, and HC [67] in vivo: IL-6 in human [68] |
Tofacitinib (Xeljanz®)FDA approved (2012) | [21][42][43][48][49][50] | [42,63 | 44] | ,64 | [45] | ,65 | [46] | ,66 | [47] | ,67 | [ | ,68,69,70,71] |
| ↑ | in vitro: IL-2 in Th1. IL-17, IL-2 in Th17 cells (polarized via TGF-β1, IL-6) [45] | : IL-2 in Th1. IL-17, IL-2 in Th17 cells (polarized via TGF-β1, IL-6) [66] | ||||||||||||||||||||
| JAK3 > JAK1, TYK2, JAK2 [42] | JAK3 > JAK1, TYK2, JAK2 [63] | ↓ | Transcription: IFN-α, IFN-β, IL-10, IL-22, IL-2, IL-4, IL-7, IL-9, IL-15, IL-2, IFN-γ > IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IL-12, IL-23, Type III IFNs, EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 in vitro: IL-4, IL-13, IFN-γ, TNF-α in PBMC after TCR stimulation, IL-4, IL-13, IFN-γ, TNF-α, IL-17A, GM-CSF in PBMC after IL-2 stimulation [51] | : IFN-α, IFN-β, IL-10, IL-22, IL-2, IL-4, IL-7, IL-9, IL-15, IL-2, IFN-γ > IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IL-12, IL-23, Type III IFNs, EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 in vitro: IL-4, IL-13, IFN-γ, TNF-α in PBMC after TCR stimulation, IL-4, IL-13, IFN-γ, TNF-α, IL-17A, GM-CSF in PBMC after IL-2 stimulation [72] |
Peficitinib (Smyraf®) Japan Approved (2019) | [42][51] | [63,72] | |||||||||||||||
| JAK2, JAK1 > TYK2 > JAK3 [42] | JAK2, JAK1 > TYK2 > JAK3 [63] | ↓ | Transcription: IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IFN-α, IFN-β, IL-10, IL-22, IFN-γ > IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 > IL-12, IL-23, Type III IFNs, EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 [43] in vitro: IL-6 in MoDCs, IFN-α secreted pDCs [44] MCP-1 [21] IL-17 in CD4+ T cells (AS, PSA, RA, and HC) [46] | : IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IFN-α, IFN-β, IL-10, IL-22, IFN-γ > IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 > IL-12, IL-23, Type III IFNs, EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 [64] in vitro: IL-6 in MoDCs, IFN-α secreted pDCs [65] MCP-1 [42] IL-17 in CD4+ T cells (AS, PSA, RA, and HC) [67] |
Baricitnib (Olumiant®) FDA approved (2018) | [21][42][43][44][46][48][49] | [42,63,64,65,67,69,70] | |||||||||||||||
| JAK2, JAK1 > TYK2 > JAK3 [42] | JAK2, JAK1 > TYK2 > JAK3 [63] | ↓ | Transcription: IFN-γ, EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 > IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IFN-α, IFN-β, IL-10, IL-22, IL-12, IL-23, Type III IFNs > IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 in vitro: IL-10, IFN-γ, IL-6, TNF-α, IL-13 [52] IL-17 in CD4+ (AS, PSA, RA, and HC) [46] in vivo: IFN-γ, IL-12p70, IL-6, G-CSF, IL-10, TNF-α [52] | : IFN-γ, EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 > IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IFN-α, IFN-β, IL-10, IL-22, IL-12, IL-23, Type III IFNs > IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 in vitro: IL-10, IFN-γ, IL-6, TNF-α, IL-13 [73] IL-17 in CD4+ (AS, PSA, RA, and HC) [67] in vivo: IFN-γ, IL-12p70, IL-6, G-CSF, IL-10, TNF-α [73] |
Ruxolitinib (Jakafi®) FDA approved (2011) (myelofibrosis) | [42][46][48][52][53] | [63,67,69,73,74] | |||||||||||||||
| ↑ | in vitro: IL-2 [53] | : IL-2 [74] | ||||||||||||||||||||
| JAK1 > JAK2 > TYK2 > JAK3 [42] | JAK1 > JAK2 > TYK2 > JAK3 [63] | ↓ | Transcription: IL-6, IL-11, IL-13, IL-25, IL-27, IL-31 > IFN-α, IFN-β, IL-10, IL-22 > IFN-γ, > IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 > EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5, IL-12, IL-23, Type III IFNs [43] in vitro: IL-2, IL-4, IFN-αB2, IFN-γ [50] IFN-α, IL-6, IFN-γ, IL-2, IL-15, IL-4 [43] ex vivo: IL-6, GM-CSF [43] ] | : IL-6 | in vivo: IFN-γ, IL-6, IL-1β, RANKL, MMP-3, MMP-13, IP10, XCL1, MCP-1, MIP-1b, MCP-3, MCP-5, M-CSF1, MDC, SCF, KC/GRO, IL-1α | , IL-11, IL-13, IL-25, IL-27, IL-31 > IFN-α, IFN-β, IL-10 | [50] SAA, IL-6, IL-1β, GM-CSF, TNF-RI, Resistin, TNF-α, MMP-3, YKL40, VEGF, MMP-1, IL-12, IL-2, IFN-γ, IL-13, IL-5, IL-21, IL-23, IL-17A, IL-7, IL-10, CXCL10, CXCL13, MCP-1, VCAM-1, MIP-1a | , IL-22 > IFN-γ, > IL-2 | [ | , IL-4, IL-7, IL-9, IL-15, IL-21 > EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5, IL-12, IL-4 | 54 | , IL-23, Type III IFNs [64] in vitro: IL-2, IFN-αB2, IFN-γ [71] IFN-α, IL-6, IFN-γ, IL-2, IL-15, IL-4 [64] ex vivo: IL-6, GM-CSF [64] in vivo: IFN-γ, IL-6, IL-1β, RANKL, MMP-3, MMP-13, IP10, XCL1, MCP-1, MIP-1b, MCP-3, MCP-5, M-CSF1, MDC, SCF, KC/GRO, IL-1α [71] SAA, IL-6, IL-1β, GM-CSF, TNF-RI, Resistin, TNF-α, MMP-3, YKL40, VEGF, MMP-1, IL-12, IL-2, IFN-γ, IL-13, IL-5, IL-21, IL-23, IL-17A, IL-7, IL-10, CXCL10, CXCL13, MCP-1, VCAM-1, MIP-1a [75] |
Filgotinib (Jyseleca®) EMA & Japan approved (2020) | [42][43][48][50][54] | [63,64,69,71,75] | |||||||
| JAK1 > JAK2 > JAK3 > TYK2 [42] | JAK1 > JAK2 > JAK3 > TYK2 [63] | ↓ | Transcription: IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IFN-α, IFN-β, IL-10, IL-22, IFN-γ EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 > IL-12, IL-23, Type III IFNs [43] in vitro: IFN-α, IL-6, IFN-γ, IL-2, IL-4, IL-15, G-CSF [43] | : IL-6, IL-11, IL-13, IL-25, IL-27, IL-31, IFN-α, IFN-β, IL-10, IL-22, IFN-γ EPO, TPO, GH, G-CSF, GM-CSF, Leptin, IL-3, IL-5 IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 > IL-12, IL-23, Type III IFNs [64] in vitro: IFN-α, IL-6, IFN-γ, IL-2, IL-4, IL-15, G-CSF [64] |
Upadacitinib (Rinvoq®) FDA approved (2019) | [42][43][49] | [63,64,70] | |||||||||||||||
| JAK2 > JAK1 > TYK2 > JAK3 [55] | JAK2 > JAK1 > TYK2 > JAK3 [76] | ↓ | See main text | in vitro: VCAM-1, IL-6 [53] | : VCAM-1, IL-6 [74] | Fedratinib (Inrebic®) (2019) (myelofibrosis) | [42][53] | 74 | [56] | ,77 | [57] | ,78 | [58] | [63,,79] | ||||||||
| ↑ | in vitro: IL-2 [53] | : IL-2 [74] |
5. Grouping of the Therapeutic Approaches
LEAPS vaccines are peptides that can be designed to elicit an antigen-directed Th1 or Th2/Treg cytokine conversation depending upon the LEAPS immune cell binding ligand peptide that is attached to a disease-related antigenic peptide [3][17][18][19][62]. The J-ICBL activates DCs which produce IL-12 to promote IFN-γ and Th1 cytokine conversations and responses whereas the DerG-ICBL acts on CD4 T cells to promote Th2 and Treg cytokine conversations and responses. By promoting the appropriate antigen-specific cytokine conversations, immunization with J-LEAPS vaccines elicit anti-viral and anti-tumor responses [63] and have the potential to modulate Th17 responses. The DerG-LEAPS vaccines elicit antibody responses [3][62] and have the potential to modulate Th1 responses.
Group6. Comparisons of LEAPS, Monoablative, and Jakinib Therapies
LEAPS peptide therapeutic vaccines are designed to have an immunomodulatory effect on the T cells driving the disease, as illustrated in Figure 23A,B. In so doing, the LEAPS peptides affect the entire cytokine conversation, increasing the expression of some and decreasing other cytokines to return immunobalance, rather than acting on a single cytokine [3][19][17][18][62][3,27,29,30,31].
The monoablative therapies (shown in Figure 23C,D) use a neutralizing monoclonal antibody or receptor-antagonist to specifically block the action of one of the disease-associated cytokines after secretion and not its synthesis. The targets for these therapies include IL-1β, IL-6, IL-17A, IL-23, IL-12, and TNF-α and, under certain circumstances, IFN-γ. These monoablation therapies only indirectly affect other pro-inflammatory cytokines and do not upregulate anti-inflammatory cytokines to rebalance the cytokine conversation.
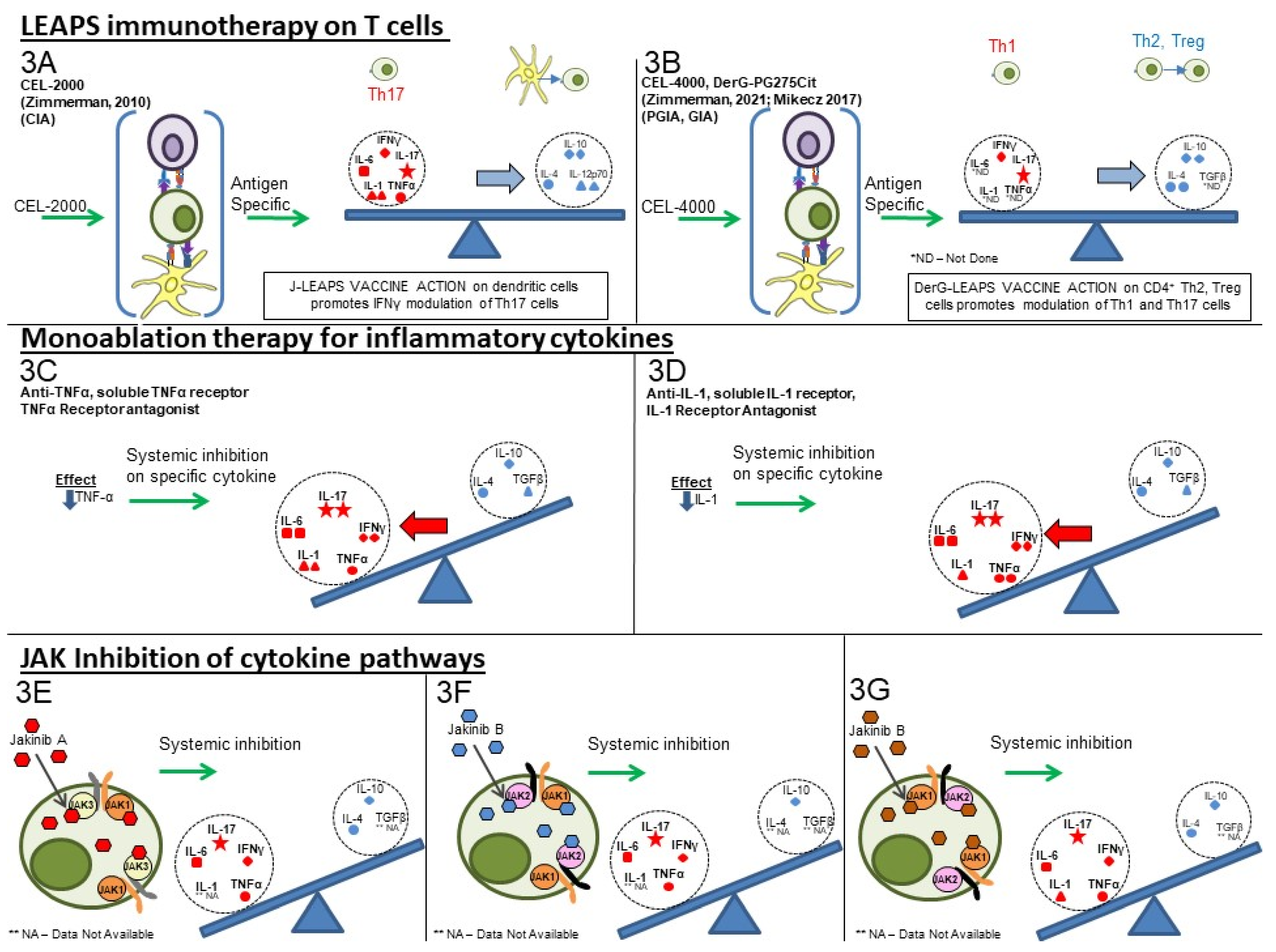
Figure 2. Comparison of cytokine-targeting therapies to treat autoimmune conditions. LEAPS immunomodulating therapy: (3A) CEL-2000 J-LEAPS vaccine: Immunization of diseased animals activates dendritic cells to promote antigen-specific

Th1 responses and IL-10 to modulate the disease driving Th17 and inflammatory cytokine responses and provide therapy. (3B) CEL-4000 and related DerG-LEAPS vaccines: CEL-4000 vaccination of diseased animals activates anti third gen-specific CD4 Th2 and Treg cells to modulate the disease driving Th1, Th17 and inflammatory cytokine responses. Treatment favors a ratio of increased anti-oup (IL-4, IL-10) vs. pro-(IFN-γ or IL-17) for cytokine secreting CD4 spleen T cells. Monoablation therapy for inflammatory cytokines (DMARDS): (3C) Neutralizing antibody to IL-1, TNFα, or IL-6 (not shown); (3D) Receptor antagonist inhibition of cytokine action: Neutralization or blocking of cytokine receptor by antibody can prevent systemic action of a specific inflammatory cytokine but also affects antimicrobial and other immune responses. Treatment has no effect on anti-inflammatory cytokines. Inhibition of JAK-tyrosine kinase cascade: (3E–3G) Inhibitors of of different JAKs: Small molecular inhibitors of JAK1, JAK2, JAK3 or tyrosine kinase 2 (TYK2) block the signal transmission from associated cytokine receptors to block inflammatory and regulatory responses, depending upon the JAKtherapies (s) that are inhibited. These inhibitors downregulate transcription of one or more cytokine gene, as listed in Table 1.
The third group (III) of therapies (Table 1, Figure 23E–G) are the jakinibs, which act by inhibiting specific receptor-associated JAK/STAT tyrosine kinases, ultimately inhibiting the synthesis and secretion of multiple cytokines (multi-ablative therapy) that are activated by the specific JAK cascade. The jakinibs are small-molecule (~300Da) inhibitors acting on the Janus kinases JAK1, JAK2, JAK3 or TYK2 and have the major advantage of being taken orally [42][63]. The JAK enzymes most often work in pairs as homo- or heterocomplexes activating STAT molecules to create transcriptional activators and promote the expression of groups of cytokines and other genes. JAK activation or inhibition also influences the expression of different cell surface receptors, including CD4, CD80 and CD86 on T, B and other cells and their associated immune responses [44][65]. The expression of some JAK enzymes is more restricted to certain cell types than others, such as JAK3 for immune system cells such as B, T, and NK cells. This is a therapeutic advantage.
As can be seen in Table 1, there are at least five different jakinibs approved for RA treatment in the USA, Europe, or Japan and several others are under investigation, each unique in terms of the molecule’s binding preference for its particular cognate JAK or JAK-associated molecule. Different manifestations of treatment occur depending on the relative selectivity of binding and whether it is reversible or irreversible. The representative jakinibs are shown as different-colored (red, blue and orange) hexagonal shapes in Figure 23 for the three examples of jakinibs that downregulate inflammatory and anti-inflammatory cytokines. Jakinib A (Figure 23E red hexagon) has activity which is JAK 3>1 and downregulates the expression of IL-6, IL-17, IFN-γ, TNF-α, IL-4, and IL-10. Jakinib B (Figure 23F blue hexagon) is a JAK 2>1 inhibitor downregulating IL-6, IL-17, IFN-γ, TNF-α, and IL-10. Jakinib C (Figure 23G orange hexagon) is a JAK 1>2 inhibitor downregulating IL-1, IL-6, IL-17, IFN-γ, TNF-α, IL-4, and IL-10. It should be noted that a jakinib specific for only JAK2 cannot be used since the inhibition of JAK2/STAT is associated with lethality early in life [42][56][63,77].
Although TNF-α, IL-1, and IL-17 are major targets for monoablation therapy, they are not directly affected by the jakinibs. However, they may be indirectly affected by the inhibition of expression of other cytokines that are supposedly not involved in the JAK signaling pathway; for example, IL-17 is affected indirectly by several of these jakinibs [45][46][56][66,67,77]. Similarly, an indirect effect of jakinibs may also promote the expression of some anti-inflammatory cytokines. The combination therapy targeting several cytokines, as possibly seen for the JAK inhibitors, may be effective, although this is still being debated and new clinical studies will be needed [2][71][72][73][2,99,111,112].