Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jia Huang Chen and Version 2 by Rita Xu.
Uremic toxins are defined as harmful metabolites that accumulate in the human body of patients whose renal function declines, especially chronic kidney disease (CKD) patients. Growing evidence demonstrates the deteriorating effect of uremic toxins on CKD progression and CKD-related complications, and removing uremic toxins in CKD has become the conventional treatment in the clinic.
- acute kidney injury
- protein-bound uremic toxins
- chronic kidney disease
1. Introduction
1.1. The Updated ADQI Consensus for AKI, AKD, and CKD
End-stage renal disease (ESRD) has had a high disease burden recently, which might be because the kidney itself is a vulnerable organ that easily suffers from numerous pathogenic insults, including nephrotoxic agents, hypoperfusion, metabolic disturbances, uremic toxins retention, and natural aging-related processes. Once an acute kidney injury (AKI) is encountered, the insults frequently lead to subsequence chronic kidney disease (CKD) progression despite a temporary recovery from AKI [1]. On the other hand, cumulative pieces of evidence also suggest that CKD acts as a predisposing factor for newly developed AKI [2]. In order to develop the consensus for AKI, acute kidney disease (AKD), and CKD, Acute Dialysis Quality Initiative (ADQI)16 announces the expert’s meetings and makes a new statement (Figure 1) [3].AKI can be demonstrated as a continuum, while initial kidney damage can lead to later renal injury and eventually lead to CKD. For the definition of AKI, it encounters an abrupt reduction in renal function occurring over 7 days or less. For CKD, it is defined by the persistence of kidney disease for a period of >90 days. Between the two clinical situations, there is a transition status which is named AKD. It describes acute or subacute damage and/or loss of kidney function for a duration of between 7 and 90 days after exposure to an AKI initiating insults. Based on clinical observation, recovery from AKI within 48 h of the initiating event typically heralds rapid recovery from AKI. For patients with pre-existing CKD, the AKI event also can be superimposed on CKD, with AKD existing on a background of CKD. AKD patients with pre-existing CKD have shown a high risk of kidney progression and future ESRD development [4].
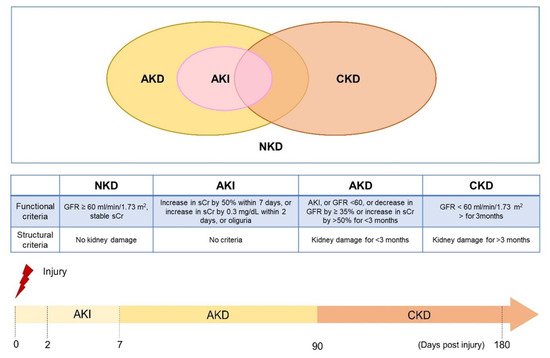
Figure 1. Definitions of kidney disease and the progression timeline (The concepts of the figure were modified from Acute Disease Quality Initiative (ADQI) 16 Workgroup).
1.2. AKI Increases the Long-Term Risk of Progressive CKD
The development of AKI is associated with increased morbidity, length of hospital stay, higher hospital costs, economic burden, and increased mortality [5][6][5,6]. Even small changes in serum creatinine (sCr) of 0.3–0.5 mg/dL are associated with increased long-term mortality [7]. Recent awareness of the prospect that a portion of patients suffering from AKI might progress to CKD or ESRD requiring maintenance dialysis, with the incidence rate of 4.9 per 100 patients/year [8][9][8,9]. According to the 2015 United States Renal Data System (USRDS) report, CKD status changes significantly in the year following an AKI hospitalization. Among patients without baseline CKD, nearly 30% are reclassified as having some degree of CKD, including 0.20% being declared ESRD [10]. Whether the patient progresses to CKD after recovery from AKI (also termed as AKI to CKD transition) and the precise mechanisms of AKI to CKD transition are still under exploration. The possible explanation of AKI to CKD transition may be that the maladaptive repair of either irreversible or sustained cellular injury, which further induces glomerulosclerosis, tubulointerstitial fibrosis, or vascular rarefaction. After suffering from AKI, uremic solutes such as the small molecule uremic toxin sCr and blood urea nitrogen (BUN) are increased and have been suggested as an indicator for kidney injury. However, it was gradually recognized that these tests were neither specific nor sensitive in several clinical practices [11][12][11,12].
2. Etiology and Pathophysiological Roles of Uremic Toxin
Uremic toxins are metabolites generated from daily food intake and their excretion into the urine through glomerular filtration or active transport by the renal proximal epithelial cell [13][15]. The accumulation of uremic toxins is characteristic of CKD development and renal function decline [14][16]. The European Uremic Toxin Work Group (EUTox) documented that uremic toxins are classified into three catalogs based on their physicochemical characteristic: water-soluble small molecule (<500 Da), middle molecule (>500 Da), and protein-bound uremic toxin [13][15]. Water-soluble small molecule uremic toxins are hydrophilic and easily penetrate from the glomerular barrier and are eliminated. Creatinine and urea are the most representative of two small-molecule uremic toxins and are widely used as surrogate markers for identifying renal function in the clinic [15][17]. Middle molecule uremic toxins, such as β2-Microglobulin, FGF23, and pro-inflammatory cytokines IL-1β, IL-6, have evolved mainly to be synonymous with peptides and proteins that accumulate in uremia [16][18]. The PBUTs, IS and PCS are two of the notorious uremic toxins, which are derived from the amino acid tryptophan and tyrosine, respectively, and are metabolised in the liver separately [17][19]. Due to their protein-bound characteristic, very little is filtrated through the glomerular barrier and they have a low clearance rate in patients undergoing hemodialysis or peritoneal dialysis [18][20]. Therefore, they have more severe adverse effects in patients with renal function impairment, and most studies are focus on exploring their deteriorating effects in CKD, cardiovascular disease, and uremic sarcopenia progression [19][20][21][22][13,21,22,23].
3. The Retention of PBUTs in Kidney Disease Models
There are many kidney disease models in uremic toxin research, whether in AKI, CKD, or AKI to CKD. Here, we listed the commonly used animal model in those studies. For AKI studies: Bilateral ischemia-reperfusion injury, cisplatin, and contrast-induced kidney injury model; For CKD studies: 5/6 nephrectomy, adenine diet, and folic acid injection; For AKI to CKD studies: UIRI, unilateral two-stage IRI. Other models such as aristolochic acid and unilateral ureteral obstruction (UUO) would be applied in AKI or CKD model based on investigator’s experimental design [23][24]. According to disease definition, both AKI and CKD models would cause impaired renal function with increasing BUN and sCr. Moreover, the renal function of the UIRI model would return to baseline within several days in the AKI model, whereas the CKD model lasted. Of note, UIRI model would not induce obvious BUN and sCr upregulation due to the compensatory effect of the contralateral kidney [24][25]. However, the injured kidney caused intensive AKI-related gene expression-havcr1 and lcn2. Even after 2–16 weeks of UIRI insult, the injured kidney formed unresolved fibrosis development and inflammatory-related genes expression, which were well-recognized features in CKD [25][26]. Regarding unilateral two-stage IRI, Wei et al. developed a novel AKI to CKD model through 2 weeks UIRI induction, then removed the contralateral intact kidney. This model presented a progressive development of AKI to CKD transition, including sustained renal function dysfunction after nephrectomy and deleterious effects in renal fibrosis [26][27]. In contrast to CKD, there are seldom studies in the field of PBUTs in AKI. AKI insult causing uremic toxin retention comes from a decline in glomerular filtration rate (GFR), leading to increased small molecule uremic toxins that cannot be eliminated through the glomerular filtrate and this alters the metabolic profiles which promote uremic toxin precursor formation. In the following section, we list the possible reason for PBUTs accumulation after AKI from many aspects, including secretory function decline, metabolic alteration, and precursor synthesis.
3.1. Loss of Secretory Function in Renal Tubular Epithelial Cells after AKI
Acute tubular necrosis is the common consequence of AKI [27][28]. With the loss of epithelial cells in renal tubules, the secretory function of kidneys are dramatically decreased after AKI due to the loss of OAT1/3 expression in proximal tubular cells [28][29]. The OATs superfamily are the important transporters expressed in RTECs and have a role in substrates reabsorption and elimination [29][30]. Among them, OAT1/3 are responsible for PBUTs transport from circulation to filtration by the tubular epitheliums and excretion. It has been found that IS accumulates in an in vivo AKI model [28][30][29,31]. Different to BUN and sCr levels which return to normal levels in the recovery stage of AKI [24][25], furthermore, our current work found that accumulation of the secreted PBUT is sustained without an increase in BUN and sCr after 10 days of unilateral ischemia-reperfusion injury mice model [31][32]. Therefore, we thought UIRI model would be a suitable model to investigate the roles of PBUTs in AKI to CKD transition.
3.2. AKI Alert Gut Microbiota Composition Leading to PBUTs Synthesis
The gut microbiota plays a crucial role in the host immune homeostasis and influence extraintestinal biologic functions. The concept of the kidney-gut axis has recently aroused more attention in the field of AKI and CKD research. Yang et al. demonstrated that ischemia reperfusion-induced AKI provokes intestinal dysbiosis, increasing Enterobacteriaceae and Escherichia coli, and decreasing Lactobacilli Ruminococacceae [32][33]. Gryp et al. reveal that a higher abundance of Enterobacteriaceae and Escherichia coli was found in CKD patients’ fecal matter [33][34], and are also responsible for PBUTs and the synthesis of precursors- p-cresol and indole [34][35][35,36]. Furthermore, the decreased levels of Lactobacillales are associated with CKD development, inflammation, short-chain fatty acid (SCFA) deficiency, and loss of intestinal permeability [36][37][37,38]. SCFAs are the metabolites of gut microbiota and serve as the energy source for intestinal epithelial cells, immune modulation, and strengthening intestinal integrity [37][38][38,39]. Since gut microbiota and its microenvironment govern PBUTs production, immune regulation, and kidney disease progression, the research about the management of gut bacterial in AKI and CKD is blooming and their therapeutic efficacy is worthy of further exploration [39][40][41][40,41,42].