A novel coronavirus (SARS-CoV-2) emerged towards the end of 2019 that caused a severe respiratory disease in humans called COVID-19. It led to a pandemic with a high rate of morbidity and mortality that is ongoing and threatening humankind. Most of the mutations occurring in SARS-CoV-2 are synonymous or deleterious, but a few of them produce improved viral functions. The first known mutation associated with higher transmissibility, D614G, was detected in early 2020. Since then, the virus has evolved; new mutations have occurred, and many variants have been described. Depending on the genes affected and the location of the mutations, they could provide altered infectivity, transmissibility, or immune escape. To date, mutations that cause variations in the SARS-CoV-2 spike protein have been among the most studied because of the protein’s role in the initial virus–cell contact and because it is the most variable region in the virus genome. Some concerning mutations associated with an impact on viral fitness have been described in the Spike protein, such as D614G, N501Y, E484K, K417N/T, L452R, and P681R, among others. To understand the impact of the infectivity and antigenicity of the virus, the mutation landscape of SARS-CoV-2 has been under constant global scrutiny. The virus variants are defined according to their origin, their genetic profile (some characteristic mutations prevalent in the lineage), and the severity of the disease they produce, which determines the level of concern. If they increase fitness, new variants can outcompete others in the population. The Alpha variant was more transmissible than previous versions and quickly spread globally. The Beta and Gamma variants accumulated mutations that partially escape the immune defenses and affect the effectiveness of vaccines. Nowadays, the Delta variant, identified around March 2021, has spread and displaced the other variants, becoming the most concerning of all lineages that have emerged. The Delta variant has a particular genetic profile, bearing unique mutations, such as T478K in the spike protein and M203R in the nucleocapsid. This entreviewy summarizes the current knowledge of the different mutations that have appeared in SARS-CoV-2, mainly on the spike protein. It analyzes their impact on the protein function and, subsequently, on the level of concern of different variants and their importance in the ongoing pandemic.
- SARS-CoV-2 genome
- Spike protein
- receptor binding domain (RBD)
- escape mutation
- neutralizing antibodies (nAbs)
- variant of concern (VOC)
- COVID-19 vaccines
1. Introduction
1.1. SARS-CoV-2 Structure
|
Gene |
Transcript |
Protein Name(s) |
Position in the Genome |
Length (aa) |
Function |
|---|---|---|---|---|---|
|
5′UTR |
1–265 |
||||
|
ORF1a |
pp1ab, pp1a [10] |
nsp1 |
266–805 |
180 |
Leader protein. Cellular mRNA degradation, inhibiting IFN signaling [2]. |
|
nsp2 |
806–2719 |
638 |
Unknown. |
||
|
nsp3, PLpro |
2720–8554 |
1945 |
Papain-like protease, adenosine diphosphate-ribose 1″-phosphatase. Blocks host innate immune response, promotes cytokine expression [2]. |
||
|
nsp4 |
8555–10054 |
500 |
Double-membrane vesicles formation [2]. |
||
|
nsp5, 3CLpro, Mpro |
10055–10972 |
306 |
3-chymotrypsin-like Cys protease. Main protease. Mediates cleavages downstream of nsp4. Inhibits IFN signaling [2]. |
||
|
nsp6 |
10973–11842 |
290 |
Restricting autophagosome expansion. Double-membrane vesicle formation [2]. |
||
|
nsp7 |
11843–12091 |
83 |
Cofactor with nsp8 and nsp12 [2]. |
||
|
nsp8 |
12092–12685 |
198 |
Replicase. Cofactor with nsp7 and nsp12. Primase [2]. |
||
|
nsp9 |
12686–13024 |
113 |
Replicase. ssRNA-binding protein. Dimerization and RNA binding [2]. |
||
|
nsp10 |
13025–13441 |
139 |
RNA synthesis protein. Scaffold and cooperation with nsp14 ExoN and nsp16 in methyltransferase activities [2,12,13,14,15][2][12][13][14][15]. |
||
|
ORF1ab |
pp1ab [10] |
nsp11 |
13442–13480 |
13 |
Endoribonuclease and 3′-to-5′ exonuclease [2]. |
|
nsp12, RdRP |
13442–16236 |
932 |
RNA-dependent RNA polymerase: replication and transcription of the viral genome. Primer dependent RdRp [2]. |
||
|
nsp13 |
16237–18039 |
601 |
DNA and RNA helicase/NTPase, 2′-O-ribose methyltransferase. RNA 5′-triphosphatase. RNA helicase 5′ triphosphatase [2]. |
||
|
nsp14 |
18040–19620 |
527 |
ExoN. 3′-to-5′ exonuclease. N7-guanine methyltransferase [2,11,12,15][2][11][12][15]. |
||
|
nsp15 |
19621–20658 |
346 |
Endoribonuclease, 3′-to-5′ exonuclease. NendoU. Evasion of dsRNA sensors [2]. |
||
|
nsp16 |
20659–21552 |
298 |
2′-O-ribose methyltransferase [16,17,18][16][17][18]. Avoids MDA5 recognition, negatively regulating innate immunity [2]. |
||
|
S |
Spike (S) |
21563–25384 |
1273 |
Structural protein; surface glycoprotein. Mediates virus–host cell binding. |
|
|
ORF3a |
ORF3a |
25393–26220 |
275 |
Ion channel activity (viroporin) activates the NLRP3 inflammasome. May play a role in virus replication and pathogenesis. |
|
|
E |
Envelope (E) |
26245–26472 |
75 |
Structural protein. Envelope protein. Facilitates assembly and release of the virus. It has ion channel activity required for pathogenesis. |
|
|
M |
Membrane (M) |
26523–27191 |
222 |
Structural protein. Membrane glycoprotein. Located in the transmembrane domain; it is the most abundant structural protein. |
|
|
ORF6 |
ORF6 |
27202–27387 |
61 |
Suppression of both primary interferon production and interferon signaling [19]. |
|
|
ORF7a |
ORF7a |
27394–27759 |
121 |
Type I transmembrane protein. |
|
|
ORF7b |
ORF7b |
27756–27887 |
43 |
Localize to the Golgi compartment. |
|
|
ORF8 |
ORF8 |
27894-28259 |
121 |
Interferes with host antiviral mechanisms [20]. |
|
|
N |
Nucleocapsid (N) |
28274–29533 |
419 |
Structural protein. Nucleocapsid phosphoprotein protects the viral RNA genome and is involved in packaging RNA into virus particles. |
|
|
ORF10 |
ORF10 |
29558–29674 |
38 |
Unknown. No transcripts identified [10]. |
|
|
3′UTR |
29675–29903 |
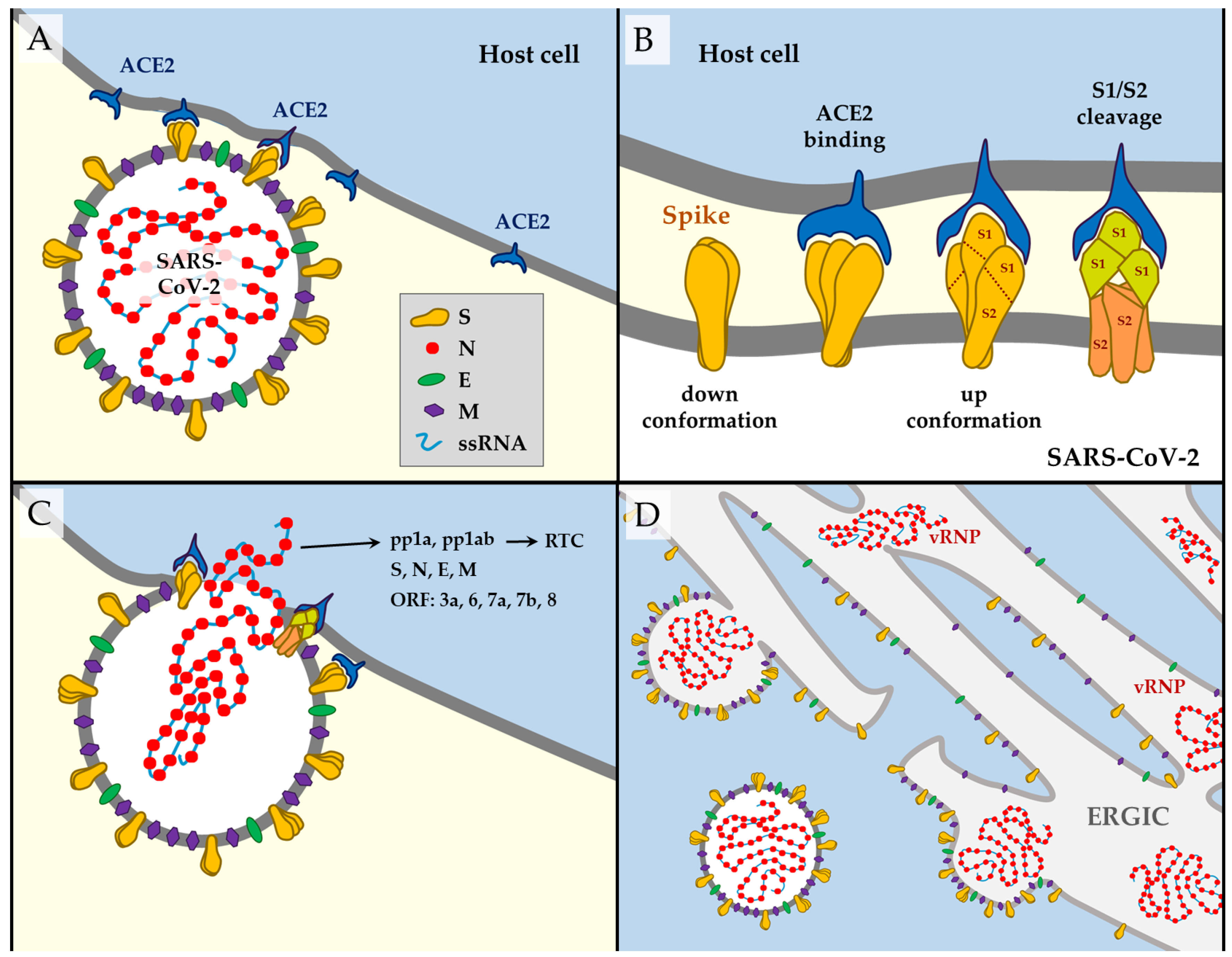
1.2. SARS-CoV-2 Way of Action
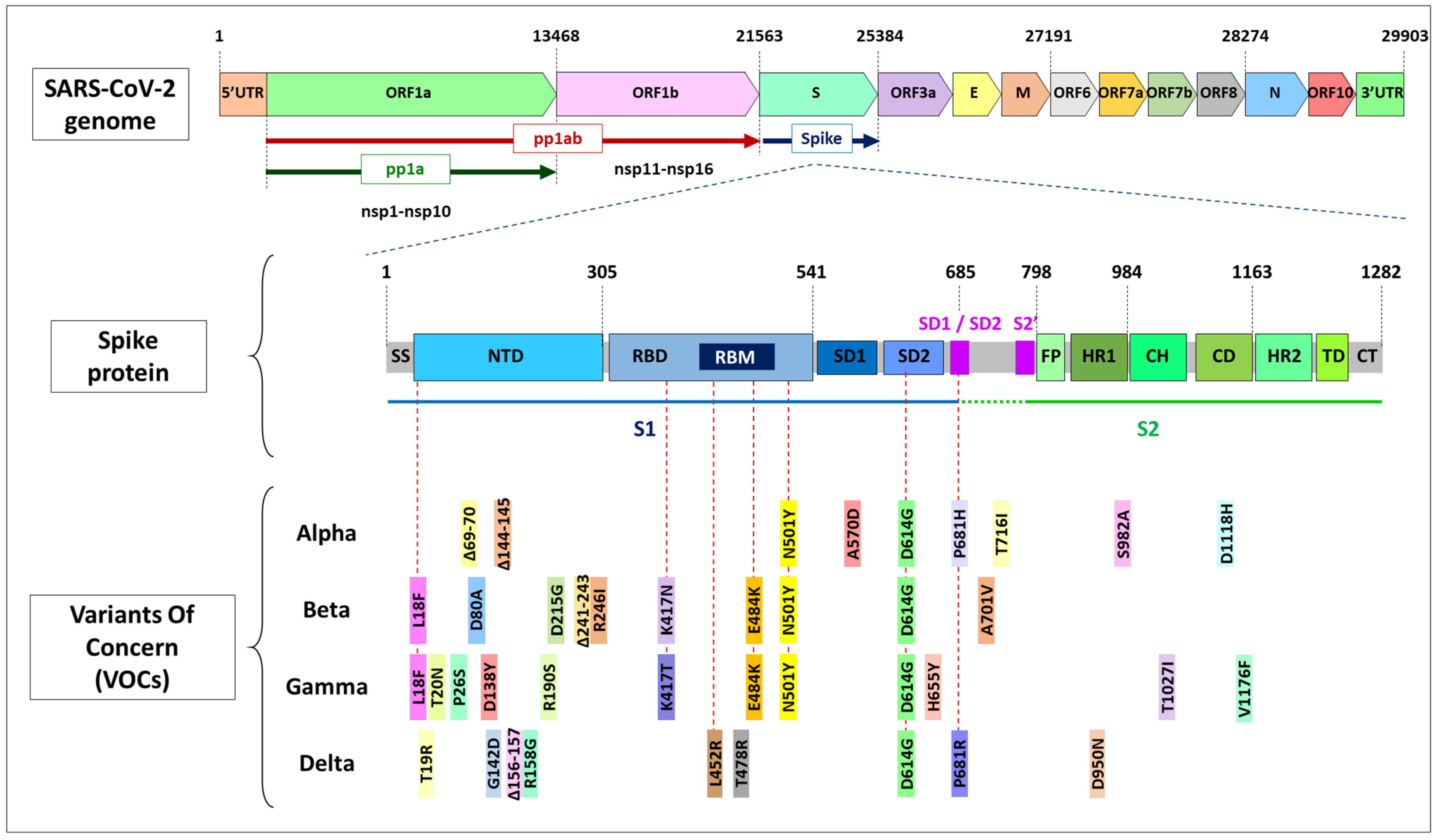
1.3. Mutations in Coronavirus ssRNA
2. Mutations in the spike Gene
2.1. D614G
2.2. N501Y
2.3. E484K
2.4. Other RBD Mutations
2.5. P681 Residue
2.6. NTD Deletions
2.7. Mutations out of the spike Gene
3. SARS-CoV-2 Lineages. Classification of Variants: VOC and VOI
4. Escape Mutations and Vaccine Efficacy
5. COVID-19 during Human Development
References
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192.
- Chen, Y.; Liu, Q.; Guo, D.J. Emerging coronaviruses: Genome structure, replication, and pathogenesis. Med. Virol. 2020, 92, 418–423.
- Salleh, M.Z.; Derrick, J.P.; Deris, Z.Z. Structural Evaluation of the Spike Glycoprotein Variants on SARS-CoV-2 Transmission and Immune Evasion. Int. J. Mol. Sci. 2021, 22, 7425.
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and charac-terization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013, 503, 535–538.
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and Avian coronaviruses as the gene source of gammacoronavirus and deltacoro-navi. J. Virol. 2012, 86, 3995–4008.
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452.
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Patho-genesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502.
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 10, 25–38.
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS- CoV-2. Gene Res. 2020, 16, 100–682.
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of sars-cov-2 transcriptome. Cell 2020, 181, 914–921.
- Bouvet, M.; Imbert, I.; Subissi, L.; Gluais, L.; Canard, B.; Decroly, E. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. USA. 2012, 109, 9372–9377.
- Ma, Y.Y.; Wu, L.J.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.N.; Lou, Z.Y.; Yan, L.M.; Zhang, R.G.; Rao, Z.H. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441.
- Chen, Y.; Su, C.; Ke, M.; Jin, X.; Xu, L.; Zhang, Z.; Wu, A.; Sun, Y.; Yang, Z.; Tien, P.; et al. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2’-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011, 7, e1002294.
- Decroly, E.; Debarnot, C.; Ferron, F.; Bouvet, M.; Coutard, B.; Imbert, I.; Gluais, L.; Papageorgiou, N.; Sharff, A.; Bricogne, G.; et al. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2’-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 2011, 7, e1002059.
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489.
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279.
- Ogando, N.; Ferron, F.; Decroly, E.; Canard, B.; Posthuma, C.C.; Snijder, E.J. The Curious Case of the Nidovirus Exoribonuclease: Its Role in RNA Synthesis and Replication Fidelity. Front. Microbiol. 2019, 10, 1813.
- Smith, E.C.; Denison, M.R. Implications of altered replication fidelity on the evolution and pathogenesis of coronaviruses. Curr. Opin. Virol. 2012, 2, 519–524.
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428.
- Zinzula, L. Lost in deletion: The enigmatic ORF8 protein of SARS-CoV-2. Biochem. Biophys. Res. Commun. 2021, 538, 116–124.
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263.
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273.
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 Spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208.
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885.
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9.
- Kalathiya, U.; Padariya, M.; Mayordomo, M.; Lisowska, M.; Nicholson, J.; Singh, A.; Baginski, M.; Fahraeus, R.; Carragher, N.; Ball, K.; et al. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. J. Clin. Med. 2020, 9, 1473.
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 and Novel Therapeutics against Corona-Virus (COVID-19); StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45.
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8.
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS- CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569.
- Nishima, W.; Kulik, M. Full-Length Computational Model of the SARS-CoV-2 Spike Protein and Its Implications for a Viral Membrane Fusion Mechanism. Viruses 2021, 13, 1126.
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129.
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.-J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162.
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876.
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of natural mutations of SARS-CoV-2 on Spike structure, conformation, and antigenicity. Science 2021, 373, eabi6226.
- Watanabe, R.; Matsuyama, S.; Shirato, K.; Maejima, M.; Fukushi, S.; Morikawa, S.; Taguchi, F. Entry from the Cell Surface of Severe Acute Respiratory Syndrome Coronavirus with Cleaved S Protein as Revealed by Pseudotype Virus Bearing Cleaved S Protein. J. Virol. 2008, 82, 11985–11991.
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620.
- Giovanetti, M.; Benedetti, F.; Campisi, G.; Ciccozzi, A.; Fabris, S.; Ceccarelli, G.; Tambone, V.; Caruso, A.; Angeletti, S.; Zella, D.; et al. Evolution patterns of SARS-CoV-2: Snapshot on its genome variants. Biochem. Biophys. Res. Commun. 2021, 538, 88–91.
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069.
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748.
- Moya, A.; Elena, S.; Bracho, M.A.; Miralles, R.; Barrio, E. The evolution of RNA viruses: A population genetics view. Proc. Natl. Acad. Sci. USA 2000, 97, 6967–6973.
- Lynch, M.; Ackerman, M.; Gout, J.-F.; Long, H.; Sung, W.; Thomas, W.K.; Foster, P.L. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016, 17, 704–714.
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19.
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. SARS-CoV-2 (COVID-19) by the numbers. eLife 2020, 9, e57309.
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell. 2020, 78, 779–784.e5.
- Sempowski, G.D.; Saunders, K.O.; Acharya, P.; Wiehe, K.J.; Haynes, B.F. Pandemic Preparedness: Developing Vaccines and Therapeutic Antibodies For COVID-19. Cell 2020, 181, 1458–1463.
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571.
- Piccoli, L.; Park, Y.J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping neutralizing and immunodominant sites on the SARS- CoV-2 spike receptor-binding domain by struc-ture-guided high-resolution serology. Cell 2020, 183, 1024–1042.
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS- CoV-2 spike. Nature 2020, 584, 450–456.
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20.
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220.
- Kawasuji, H.; Takegoshi, Y.; Kaneda, M.; Ueno, A.; Miyajima, Y.; Kawago, K.; Fukui, Y.; Yoshida, Y.; Kimura, M.; Yamada, H.; et al. Transmissibility of COVID-19 depends on the viral load around onset in adult and symptomatic patients. PLoS ONE 2020, 15, e0243597.
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255.
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 2021, 29, 463–476.e6.
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 Spike protein variants. eLife 2020, 9, e61312.
- Andreano, E.; Piccini, G.; Licastro, D.; Casalino, L.; Johnson, N.V.; Paciello, I.; Dal Monego, S.; Pantano, E.; Manganaro, N.; Manenti, A.; et al. SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv 2020.
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 Spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021, 29, 477–488.e4.
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6.
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 82, 1295–1310.e20.
- Winger, A.; Caspari, T. The Spike of Concern-The Novel Variants of SARS-CoV. Viruses 2021, 13, 1002.
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530.
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike Variant D614G Favors an Open Conformational State. bioRxiv 2020.
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-protein D614G mutation increases virion Spike density and infectivity. Nat. Commun. 2020, 11, 6013.
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature 2021, 592, 122–127.
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121.
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11.
- Shi, P.Y.; Plante, J.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.; Lokugamage, K.; Zhang, X.; Muruato, A.; Zou, J.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness and Neutralization Susceptibility. Nature 2021, 592, 116–121.
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poz-nansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468.
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of Spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. eLife 2021, 10, e69091.
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y Spike sub-stitution enhances SARS-CoV-2 transmission. bioRxiv 2021.
- Xie, X.; Zou, J.; Fontes-Garfias, C.; Xia, H.; Swanson, K.; Cutler, M.; Cooper, D.; Menachery, V.D.; Weaver, S.; Dormitzer, P.R.; et al. Neutralization of N501Y mutant SARS-CoV-2 by BNT162b2 vaccine-elicited sera. Res. Sq. 2021, 3, rs-143532.
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055.
- Nextstrain. Genomic Epidemiology of Novel Coronavirus—Global subsampling. Available online: https://nextstrain.org/ncov/global (accessed on 15 September 2021).
- Ku, Z.; Xie, X.; Davidson, E.; Ye, X.; Su, H.; Menachery, V.D.; Li, Y.; Yuan, Z.; Zhang, X.; Muruato, A.E.; et al. Molecular determinants and mechanism for antibody cocktail preventing SARS-CoV-2 escape. Nat. Commun. 2021, 12, 469.
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; García-Sastre, A.; Schotsaert, M. SARS-CoV-2 Spike E484K mutation reduces antibody neutralisation. Lancet Microbe. 2021, 2, e283–e284.
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 Spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018.
- Diamond, M.; Chen, R.; Xie, X.; Case, J.; Zhang, X.; VanBlargan, L.; Liu, Y.; Liu, J.; Errico, J.; Winkler, E.; et al. SARS-CoV-2 variants show resistance to neutralization by many monoclonal and serum-derived polyclonal antibodies. Res. Sq. 2021, 3, 228079.
- ECDC. Risk Related to Spread of New SARSCoV-2 Variants of Concern in the EU/EEA (2020). Available online: https://www.ecdc.europa.eu/sites/default/files/documents/COVID-19-risk-related-to-spread-of-new-SARS-CoV-2-variants-EU-EEA.pdf (accessed on 15 June 2021).
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269.
- Andreano, E.; Piccini, G.; Licastro, D.; Casalino, L.; Johnson, N.V.; Paciello, I.; Dal Monego, S.; Pantano, E.; Manganaro, N.; Manenti, A.; et al. SARS-CoV-2 escape from a highly neutralizing COVID-19 convalescent plasma. Proc. Natl. Acad. Sci. USA 2021, 118, e2103154118.
- Callaway, E. Could new COVID variants undermine vaccines? Labs scramble to find out. Nature 2021, 589, 177–178.
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854.
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687.
- Tchesnokova, V.; Kulasekara, H.; Larson, L.; Bowers, V.; Rechkina, E.; Kisiela, D.; Sledneva, Y.; Choudhury, D.; Maslova, I.; Deng, K.; et al. Acquisition of the L452R mutation in the ACE2-binding interface of Spike protein triggers recent massive expansion of SARS-CoV-2 variants. J. Clin. Microbiol. 2021, 59, e0092121.
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654.
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in hu-manized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014.
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Landscape analysis of escape variants identifies SARS-CoV-2 Spike mutations that attenuate monoclonal and serum antibody neutralization. bioRxiv 2021.
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico’, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 Spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021, 184, 1171–1187.e20.
- Treanor, J. Influenza Vaccine—Outmaneuvering Antigenic Shift and Drift. N. Engl. J. Med. 2004, 350, 218–220.
- Chen, C.; Boorla, V.S.; Banerjee, D.; Chowdhury, R.; Cavener, V.S.; Nissly, R.H.; Gontu, A.; Boyle, N.R.; Vandergrift, K.; Nair, M.S.; et al. Computational prediction of the effect of amino acid changes on the binding affinity between SARS-CoV-2 spike protein and the human ACE2 receptor. Proc. Natl. Acad. Sci. USA 2021, 118, e2106480118.
- Gan, H.H.; Twaddle, A.; Marchand, B.; Gunsalus, K.C. Structural modeling of the SARS-CoV-2 spike/human ACE2 complex interface can identify high-affinity variants associated with increased transmissibility. J. Mol. Biol. 2021, 433, 167051.
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625.
- Cele, S.; Network for Genomic Surveillance in South Africa; Gazy, I.; Jackson, L.; Hwa, S.-H.; Tegally, H.; Lustig, G.; Giandhari, J.; Pillay, S.; Wilkinson, E.; et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature 2021, 593, 142–146.
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622.
- Follis, K.E.; York, J.; Nunberg, J.H. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 2006, 350, 358–369.
- Lau, S.Y.; Wang, P.; Mok, B.W.; Zhang, A.J.; Chu, H.; Lee, A.C.; Deng, S.; Chen, P.; Chan, K.H.; Song, W.; et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg. Microbes Infect. 2020, 9, 837–842.
- Postnikova, O.A.; Uppal, S.; Huang, W.; Kane, M.A.; Villasmil, R.; Rogozin, I.B.; Poliakov, E.; Redmond, T.M. The Functional Consequences of the Novel Ribosomal Pausing Site in SARS-CoV-2 Spike Glycoprotein RNA. Int. J. Mol. Sci. 2021, 22, 6490.
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M.; Rapp, M.; Reddem, E.R.; Yu, J.; Bahna, F.; Bimela, J.; et al. Potent SARS-CoV-2 neutralizing antibodies directed against Spike N-terminal domain target a single supersite. Cell Host Microbe 2021, 29, 819–833.
- McCallum, M.; Marco, A.; Lempp, F.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. bioRxiv 2021.
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282.
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 Spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142.
- Voss, W.N.; Hou, Y.J.; Johnson, N.V.; Delidakis, G.; Kim, J.E.; Javanmardi, K.; Horton, A.P.; Bartzoka, F.; Paresi, C.J.; Tanno, Y.; et al. Prevalent, protective, and convergent IgG recognition of SARS-CoV-2 non-RBD Spike epitopes. Science 2021, 372, 1108–1112.
- Suryadevara, N.; Shrihari, S.; Gilchuk, P.; VanBlargan, L.A.; Binshtein, E.; Zost, S.J.; Nargi, R.S.; Sutton, R.E.; Winkler, E.S.; Chen, E.C.; et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 Spike protein. Cell 2021, 184, 2316–2331.e15.
- Bal, A.; Destras, G.; Gaymard, A.; Stefic, K.; Marlet, J.; Eymieux, S.; Regue, H.; Semanas, Q.; d’Aubarede, C.; Billaud, G.; et al. Two-step strategy for the identification of SARS-CoV-2 variant of concern 202012/01 and other variants with Spike deletion H69-V70, France, August to December 2020. COVID-Diagnosis HCL Study Group. Euro Surveill. 2021, 26, 2100008.
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 Spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292.
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260.
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 14004.
- Benedetti, F.; Snyder, G.A.; Giovanetti, M.; Angeletti, S.; Gallo, R.C.; Ciccozzi, M.; Zella, D. Emerging of a SARS-CoV-2 viral strain with a deletion in nsp1. J. Transl. Med. 2020, 18, 3–29.
- Jauregui, A.R.; Savalia, D.; Lowry, V.K.; Farrell, C.M.; Wathelet, M.G. Identification of residues of SARS-CoV nsp1 that differ-entially affect inhibition of gene expression and antiviral signaling. PLoS ONE 2013, 29, 62–416.
- Holland, L.A. An 81-nucleotide deletion in SARS-CoV-2 ORF7a identified from sentinel surveillance in Arizona (january to March 2020). J. Virol. 2020, 20, 84–94.
- Bal, A. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clin. Microbiol. Infect. 2020, 26, 960–962.
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407.
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023.
- Srivastava, S.; Banu, S.; Singh, P.; Sowpati, D.T.; Mishra, R.K. SARS-CoV-2 genomics: An Indian perspective on sequencing viral variants. J. Biosci. 2021, 46, 22.
- Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions (2021). Available online: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html (accessed on 15 September 2021).
- European Centre for Disease Prevention and Control. SARS-CoV-2 Variants of Concern as of 13 September 2021. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 15 September 2021).
- World Health Organization. Tracking SARS-CoV-2 Variants. 2021. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 15 September 2021).
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.H.; Boucau, J.; Bowman, K.; et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N. Engl. J. Med. 2020, 383, 2291–2293.
- Avanzato, V.A.; Matson, M.J.; Seifert, S.N.; Pryce, R.; Williamson, B.N.; Anzick, S.L.; Barbian, K.; Judson, S.D.; Fischer, E.R.; Martens, C.; et al. Case study: Prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell 2020, 183, 1901–1912.e9.
- Naveca, F.; da Costa, C.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Costa, Á.; Duarte, D.; Silva, G.; Mejía, M.; et al. SARS-CoV-2 Reinfection by the New Variant of Concern (VOC) P.1 in Amazonas, Brazil. 2021. Available online: https://virological.org/t/sars-cov-2-reinfection-by-the-new-variant-of-concern-voc-p-1-in-amazonas-brazil/596 (accessed on 20 June 2021).
- Hoffmann, M.; Hofmann-Winkler, H.; Krüger, N.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Arora, P.; Sidarovich, A.; Moldenhauer, A.-S.; Winkler, M.S.; et al. SARS-CoV-2 variant B.1.617 is resistant to bamlanivimab and evades antibodies induced by infection and vaccination. Cell Rep. 2021, 36, 109415.
- World Health Organization. WHO Announces Simple, Easy-to-Say Labels for SARS-CoV-2 Variants of Interest and Concern. Available online: https://www.who.int/news/item/31-05-2021-who-announces-simple-easy-to-say-labels-for-sars-cov-2-variants-of-interest-and-concern (accessed on 15 June 2021).
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison’, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424.
- Li, D.-D.; Li, Q.-H. SARS-CoV-2: Vaccines in the pandemic era. Mil. Med. Res. 2021, 8, 1–15.
- Tregoning, J.S.; Brown, E.S.; Cheeseman, H.M.; Flight, K.E.; Higham, S.L.; Lemm, N.M.; Pierce, B.F.; Stirling, D.C.; Wang, Z.; Pollock, K.M. Vaccines for COVID-19. Clin. Exp. Immunol. 2020, 202, 162–192.
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655.
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Oxford COVID Vaccine Trial Group Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478.
- Bos, R.; Rutten, L.; van der Lubbe, J.E.M.; Bakkers, M.J.G.; Hardenberg, G.; Wegmann, F.; Zuijdgeest, D.; de Wilde, A.H.; Koornneef, A.; Verwilligen, A.; et al. Ad26 vector-based COVID-19 vaccine encoding a prefusion-stabilized SARS-CoV-2 Spike immunogen induces potent humoral and cellular immune responses. NPJ Vaccines 2020, 5, 91.
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell 2021, 184, 2939–2954.e9.
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615.
- Liu, Y.; Liu, J.; Xia, H.; Zhang, X.; Fontes-Garfias, C.R.; Swanson, K.A.; Cai, H.; Sarkar, R.; Chen, W.; Cutler, M.; et al. Neutralizing Activity of BNT162b2-Elicited Serum. N. Engl. J. Med. 2021, 384, 1466–1468.
- Kuzmina, A.; Khalaila, Y.; Voloshin, O.; Keren-Naus, A.; Boehm-Cohen, L.; Raviv, Y.; Shemer-Avni, Y.; Rosenberg, E.; Taube, R. SARS-CoV-2 spike variants exhibit differential infectivity and neutralization resistance to convalescent or post-vaccination sera. Cell Host Microbe 2021, 29, 522–528.e2.
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726.
- Wall, E.C.; Wu, M.; Harvey, R.; Kelly, G.; Warchal, S.; Sawyer, C.; Daniels, R.; Hobson, P.; Hatipoglu, E.; Ngai, Y.; et al. Neutralising antibody activity against SARS-CoV-2 VOCs, B.1.617.2 and B.1.351 by BNT162b2 vaccination. Lancet 2021, 397, 2331–2333.
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021, 384, 1885–1898.
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416.
- Wu, K.; Werner, A.P.; Koch, M.; Choi, A.; Narayanan, E.; Stewart-Jones, G.B.; Colpitts, T.; Bennett, H.; Boyoglu-Barnum, S.; Shi, W.; et al. Serum Neutralizing Activity Elicited by mRNA-1273 Vaccine. N. Engl. J. Med. 2021, 384, 1468–1470.
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393.e12.
- Mwenda, M.; Saasa, N.; Sinyange, N.; Busby, G.; Chipimo, P.J.; Hendry, J.; Kapona, O.; Yingst, S.; Hines, J.Z.; Minchella, P.; et al. Detection of B.1.351 SARS-CoV-2 Variant Strain—Zambia, December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 280–282.
- Emary, K.R.W.; Golubchik, T.; Aley, P.K.; Ariani, C.V.; Angus, B.; Bibi, S.; Blane, B.; Bonsall, D.; Cicconi, P.; Charlton, S.; et al. Oxford COVID-19 Vaccine Trial Group. Efficacy of ChAdOx1 nCoV-19 (AZD1222) vaccine against SARS-CoV-2 variant of concern 202012/01 (B.1.1.7): An exploratory analysis of a randomised controlled trial. Lancet 2021, 397, 1351–1362.
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280.
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594.
- Liu, J.; Liu, Y.; Xia, H.; Zou, J.; Weaver, S.C.; Swanson, K.A.; Cai, H.; Cutler, M.; Cooper, D.; Muik, A.; et al. BNT162b2-elicited neu-tralization of B.1.617 and other SARS-CoV-2 variants. Nature 2021, 596, 273–275.
- Mohammadi, M.; Shayestehpour, M.; Mirzaei, H. The impact of spike mutated variants of SARS-CoV2 on the efficacy of subunit recombinant vaccines. Braz. J. Infect. Dis. 2021, 25, 101606.
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119.
- Richards, N.E.; Keshavarz, B.; Workman, L.J.; Nelson, M.R.; Platts-Mills, T.A.E.; Wilson, J.M. Comparison of SARS-CoV-2 Antibody Response by Age Among Recipients of the BNT162b2 vs the mRNA-1273 Vaccine. JAMA Netw. Open 2021, 4, e2124331.
- Puranik, A.; Lenehan, P.J.; Silvert, E.; Niesen, M.J.M.; Corchado-Garcia, J.; O’Horo, J.C.; Virk, A.; Swift, M.D.; Halamka, J.; Badley, A.D.; et al. Comparison of two highly-effective mRNA vaccines for COVID-19 during periods of Alpha and Delta variant prevalence. medRxiv 2021.
- World Health Organization. The Janssen Ad26.COV2.S COVID-19 Vaccine: What You Need to Know. 2021. Available online: https://www.who.int/news-room/feature-stories/detail/the-j-j-covid-19-vaccine-what-you-need-to-know (accessed on 15 September 2021).
- Tarke, A.; Sidney, J.; Methot, N.; Yu, E.D.; Zhang, Y.; Dan, J.M.; Goodwin, B.; Rubiro, P.; Sutherland, A.; Wang, E.; et al. Impact of SARS-CoV-2 variants on the total CD4+ and CD8+ T cell reactivity in infected or vaccinated individuals. Cell Rep. Med. 2021, 2, 100355.
- Williams, P.C.; Howard-Jones, A.R.; Hsu, P.; Palasanthiran, P.; Gray, P.E.; McMullan, B.J.; Britton, P.N.; Bartlett, A.W. SARS-CoV-2 in children: Spectrum of disease, transmission and immunopathological underpinnings. Pathology 2020, 52, 801–808.
- Villar, J.; Ariff, S.; Gunier, R.B.; Thiruvengadam, R.; Rauch, S.; Kholin, A.; Roggero, P.; Prefumo, F.; do Vale, M.S.; Cardona-Perez, J.A.; et al. Maternal and Neonatal Morbidity and Mortality among Pregnant Women with and Without COVID-19 Infection: The INTERCOVID Multinational Cohort Study. JAMA Pediatr. 2021, 175, 817–826.
- Campbell, F.; Archer, B.; Laurenson-Schafer, H.; Jinnai, Y.; Konings, F.; Batra, N.; Pavlin, B.; Vandemaele, K.; Van Kerkhove, M.D.; Jombart, T.; et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Euro Surveill. 2021, 26, 2100509.
- Adhikari, E.H.; SoRelle, J.A.; McIntire, D.D.; Spong, C.Y. Increasing severity of COVID-19 in pregnancy with Delta (B.1.617.2) variant surge. Am. J. Obstet. Gynecol. 2021.
- Wang, A.M.; Berry, M.; Moutos, C.P.; Omere, C.; Clark, S.M.; Harirah, H.M.; Jain, S.; Olson, G.L.; Pacheco, L.D.; Saade, G.R.; et al. Association of the Delta (B.1.617.2) Variant of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) With Preg-nancy Outcomes. Obstet Gynecol. 2021, 138, 838–841.
- Saavedra, L.P.J.; Prates, K.V.; Gonçalves, G.D.; Piovan, S.; Matafome, P.; Mathias, P.C.D.F. COVID-19 During Development: A Matter of Concern. Front. Cell Dev. Biol. 2021, 9, 659032.
- Fernandes, G.M.; Motta, F.; Sasaki, L.M.P.; Silva, Â.P.D.; Miranda, A.M.; Carvalho, A.O.; Gomides, A.P.M.; Soares, A.A.S.M.; Santos, A.C.M.D., Jr.; Alves, C.O.; et al. Pregnancy Outcomes and Child Development Effects of SARS-CoV-2 Infection (PROUDEST Trial): Protocol for a Multicenter, Prospective Cohort Study. JMIR Res. Protoc. 2021, 10, e26477.
- de Oliveira, A.P.; Lopes, A.L.F.; Pacheco, G.; de Sá Guimarães Nolêto, I.R.; Nicolau, L.A.D.; Medeiros, J.V.R. Premises among SARS-CoV-2, dysbiosis and diarrhea: Walking through the ACE2/mTOR/autophagy route. Med. Hypotheses. 2020, 144, 110243.