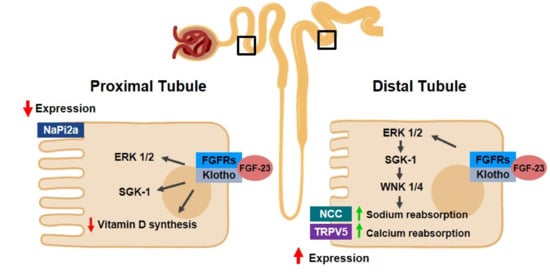
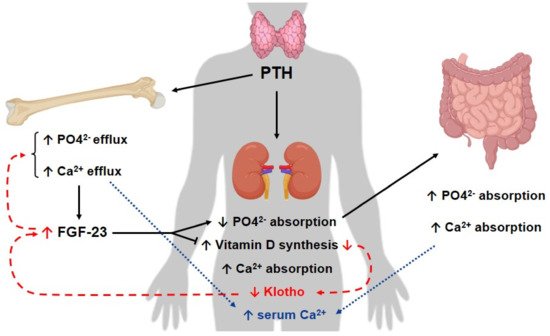
3. Acute Kidney Injury
Acute kidney injury (AKI) is a disease with a sudden onset
[103][112], marked by renal dysfunction that develops from a few hours to within seven days, according to the KDIGO Acute Kidney Injury Work Group. This disease is characterized by renal and extrarenal complications in organs such as the heart and the brain
[104][105][113,114], due to the imbalance in electrolytes
[106][115] and the accumulation of waste products
[107][108][109][116,117,118]. In the kidneys, it can vary from minor renal deterioration to ESKD, especially in patients with CKD history, leading to dialysis
[103][110][111][112,119,120]. Hence, this condition is associated with both high mortality and morbidity
[107][112][113][116,121,122].
The worldwide prevalence of AKI is increasing and prior data in the literature have shown, for instance, that individuals who survive AKI can have a 28% rate of mortality in the first year after the onset of the disease
[114][123], as well as a 50% increase in the risk of mortality during the period of approximately 10 years of follow-up
[115][116][124,125]. These patients might face other long-term outcomes, such as a higher risk of CKD
[117][126], although the exact mechanisms for this process are not yet well elucidated
[118][127]. Furthermore, patients in intensive care units (ICU) have a 50–70% rate of AKI
[107][119][120][116,128,129]; therefore, this disease is considered one of the most worrying issues for hospitalized individuals in ICUs
[108][121][122][123][117,130,131,132]. As expected, acute kidney injury also has an impact on costs in the health system
[124][125][133,134].
This syndrome has diverse etiologies, such as other prior conditions, such as sepsis, acute or chronic illnesses
[126][135], ischemia-reperfusion injury (IRI) and even the use of nephrotoxic drugs
[106][115]. Likewise, the aging of the population represents another important trend to a higher incidence of AKI
[127][136].
The standard diagnosis for AKI includes an increase in either serum creatinine of 0.3 mg/dL or more within forty-eight hours, in serum creatinine by 1.5 times within seven days or a urine volume inferior to 0.5 mL/kg/h for six hours
[108][128][117,137]. A reduction in the glomerular filtration rate (eGFR)
[129][138] is also a common consequence of this condition.
There are a variety of pathological processes associated with AKI, although the exact mechanisms involved in its physiopathology are not yet completely understood
[130][131][139,140]. Proximal tubule cells are the main cells affected after a nephrotoxic insult
[132][141] and inflammation is an important response for the development of AKI
[133][134][135][142,143,144]. Proinflammatory molecules are released by renal and endothelial cells, resulting in an infiltration of inflammatory cells
[136][145]. As a consequence, there is damage to renal tubules, characterized by cell death via necrosis and apoptosis, cytoskeleton disruption
[106][115] and oxidative stress
[137][146], for instance. Moreover, after a severe injury in the kidneys, tubular fibrosis and a senescent-like phenotype in tubular cells
[138][147] might occur. The upregulated production of profibrotic factors, such as TGF-B, leads to the activation and proliferation of fibroblasts
[139][140][148,149], stimulating the production of extracellular matrix and tubulointerstitial inflammation
[140][141][142][149,150,151]. There are also other proinflammatory molecules that contribute to the progression of AKI, such as NF-κB
[143][144][145][152,153,154].
As previously mentioned, severe AKI is considered an independent and important risk factor for the course of CKD
[115][116][146][147][124,125,155,156]; as such, patients who experience AKI are more likely to present either CKD or ESKD
[148][149][157,158]. Likewise, patients with CKD might also have transient states of renal dysfunction corresponding to AKI
[148][157].
In general, the renal impairment caused by this disease is reversible, although long-term outcomes might exist, as previously discussed
[150][159]. In addition, AKI is associated with prolonged hospitalization. There is, however, a lack of efficient therapies for AKI currently and few biomarkers that are representative of the early stage of the disease have been used in clinical practice
[151][152][153][154][160,161,162,163]. Thus, both prevention and treatment of this condition are of pivotal importance
[106][136][115,145].
In this context, Klotho has been suggested to be possibly related to AKI, as will be addressed next.
3.1. Klotho in Acute Kidney Injury
Studies have demonstrated that Klotho production is reduced in different models of AKI, such as in cisplatin-induced AKI
[137][146] and AKI induced by IRI
[155][164], which contributes to kidney damage during this disease
[46][136][156][46,145,165]. Hu et al. reported, for instance, in a preclinical and clinical study, that in rodents with IRI-induced AKI, the levels of Klotho were reduced in the kidneys, urine and blood. Moreover, they also showed that a decrease in this protein level occurred before the reduction of other early biomarkers for kidney injury
[46]. In AKI patients, researchers also detected a reduction of urinary Klotho levels, compared to healthy individuals
[46].
In order to evaluate the role of Klotho in AKI, the same research group induced different levels of this protein in mice. A higher resistance to injury was found in rodents with a higher expression of Klotho; hence, these animals displayed fewer kidney alterations, which indicates that the overexpression of Klotho might mitigate AKI, whereas its deficiency accentuates the disease
[46]. Concerning the restoration of Klotho levels, in rats, the administration of recombinant Klotho led to less renal damage in comparison to the group with no such treatment. Furthermore, it has been shown that the earlier the injection of Klotho after ischemia, the more effective this approach is to improve kidney conditions in AKI
[46]. Other studies conducted with animal models also point out the relevance of this strategy in improving renal fibrosis and pathogenesis of AKI
[137][146].
Taken together, these results provide evidence that there is indeed a deficiency of Klotho in AKI and that this contributes to renal damage. Moreover, they also highlight that this protein is both an early biomarker
[38][46][38,46] and a contributor to AKI pathogenesis
[137][146], and it is thus possible to study it as a potential therapeutic tool, considering that renal injuries are attenuated upon administration.
The exact mechanisms through which Klotho influences AKI and is downregulated are not well elucidated yet, though; some of these will be described below. It is important to note that there are several different models for the study of AKI; cisplatin-induced renal injury is a widely accepted one
[136][145] and Klotho is reduced in this model
[155][164]. This protein level, however, is also reduced in other models of AKI, such as in IRI
[46][137][46,146], AKI induced by LPS and folic acid
[157][110].
3.1.1. Klotho, Inflammation and AKI
During the development and progression of AKI, there is a dysregulation in cellular processes; some of them are related to Klotho. Data have indicated that the downregulation of Klotho in AKI is associated with cellular senescence and, importantly, that this process might be induced as a response to oxidative stress
[158][166], which is a contributor to inflammation. Moreover, reports have shown that Klotho protects the kidneys, having an anti-oxidative
[159][160][167,168] role, since it can stimulate the expression of antioxidant enzymes
[161][169]. These data are supported by the fact that a deficiency of Klotho was also shown to be associated with increased levels of oxidative stress
[162][170] in IRI models of AKI. Furthermore, experiments involving H
2O
2 as an insult similar to IRI in rodents have highlighted that co-incubation of cells with Klotho reduced the release of lactate dehydrogenase (LDH)
[163][171]. It has been reported by Sun, M., et al., 2019, for instance, in a study involving septic mice with AKI, that Klotho has a renal protective role and the mechanism for this process is related to the maintenance of mitochondrial integrity and protection against oxidative stress
[164][172].
Furthermore, Bi, F., et al., 2018, observed that Klotho is able to suppress lipopolysaccharide (LPS) AKI through the degradation—via deglycosilation—of toll-like receptor (TLR) 4
[165][173].
Regarding NF-κB in studies conducted in mice, TWEAK and TNF-α were responsible for the downregulation of Klotho, and it has been reported that the mechanism involves NF-κB
[157][110]. Furthermore, data also suggest that NF-κB is able to suppress Klotho expression through association with histone deacetylase (HDAC) 1 and nuclear receptor corepressor (NCoR), which interacts with Klotho promoters and may repress its transcription in inflammatory conditions. Moreover, in the same study, the researchers present further evidence of the importance of the anti-inflammatory effects of Klotho in AKI
[166][174]. Klotho silencing leads to an aggravated inflammatory response in a rhabdomyolysis model, causing higher expression of TNF-α and IL-1β, when compared to control mice injected with siRNA-control. Interestingly, Klotho is of pivotal importance for the renoprotective effects of nicotinamide, the active form of vitamin B3, which prevents NF-κB and corepressors recruitment to Klotho promoter, therefore attenuating inflammation and rhabdomyolysis-induced AKI and preserving Klotho expression
[166][174].
Hence, Klotho is seen as a potential anti-inflammatory molecule in AKI
[160][162][165][168,170,173], due to its association with NF-κB and its protective role against oxidative stress, for example. However, it is worth mentioning that there is a scarcity of data in the literature relating to the exact mechanisms through which Klotho is associated with inflammation in AKI, which highlights the importance of further studies regarding this topic.
3.1.2. Klotho and Non-Inflammatory Mechanisms in AKI
In addition to the inflammatory aspects mentioned above, there are also non-inflammatory events associated with Klotho and AKI.
Concerning its anti-fibrotic function, Klotho is an endogenous Wnt antagonist, blocking, as a result, the activation of β-catenin. Through the inhibition of this cascade, as previously mentioned, by binding to Wnt ligands (such as Wnt1 and Wnt4), increased levels of Klotho can reduce fibrosis in kidneys and ameliorate renal function
[100][167][100,175]. In animal models of AKI, such as unilateral ureteral obstruction, restoration of Klotho can avoid renal fibrosis
[168][169][176,177]. Prior studies have also shown that, in mice, Klotho inhibits Smad signaling induced by TGF-β; as a result, it interrupts fibrotic signaling. There are other mechanisms through which Klotho exerts an anti-fibrotic function in the kidneys, such as the inhibition of HDAC. This inhibition is causally affected by Klotho and contributes to bone morphogenetic protein 7 (BMP-7) restoration, a protein that has a renal protection role by promoting the repair and proliferation of cells from renal tubules cells after injuries
[170][178]. The downregulation of BMP-7 worsens renal complications
[171][172][179,180]. Furthermore, data from an UUO mice model of AKI demonstrated that the administration of soluble Klotho was able to suppress fibrosis, through binding to the type II receptor of TGF-β, which inhibits TGF-β signaling
[169][177].
Moreover, studies have indicated that there is cycle arrest in AKI
[138][147]. Researchers have demonstrated a positive correlation between G2/M cell cycle arrest and the synthesis of cytokines related to the fibrotic process in tubular cells, through a c-jun NH(2)-terminal kinase (JNK) signaling pathway
[138][147]. Cell cycle arrest may also lead cells to senescence. Klotho, in turn, was shown to be protective against cell senescence after AKI induction
[173][181]. Studies have shown, for instance, the attenuation of apoptosis and senescence in cultured endothelial cells after Klotho recombinant protein administration, through mitogen activated-kinase kinase (MAPK) and ERK signaling pathways
[173][181]. Furthermore, in vivo studies with mice show that the overexpression of Klotho can abrogate senescence phenotypes in injured renal tissue. In animals with a high expression of Klotho, there is also a reduction in mitochondrial DNA damage, which has been attributed by researchers to this protein. Moreover, researchers have observed a decrease in oxidative stress when Klotho is overexpressed
[47].
There is also evidence of increased Wnt signaling pathway activation in animal models that are deficient in Klotho. This event is associated, in vitro and in vivo, with cellular senescence
[174][182]. Thus, Klotho has been associated with the inhibition of the Wnt/β-pathway in AKI, and it has therefore been considered as an anti-fibrotic molecule.
In mouse models for AKI induced through IRI, in turn, studies have demonstrated that the delivery of Klotho leads to improvements in apoptosis, histological damage and creatinine values. It has been reported that there is an increase in HSP (heat shock protein) 7o expression according to Klotho levels in this animal model, which contributes to the amelioration of apoptosis
[175][183]. Hence, Klotho has also been shown to be involved with the reduction of both senescence and apoptosis, along with the improvement of renal parameters, in different models of study involving AKI.
Moreover, autophagy is activated in several models of AKI, such as in unilateral ureteral obstruction
[176][177][184,185], IRI
[178][179][186,187] and cisplatin-induced AKI
[180][181][182][183][184][188,189,190,191,192], and different studies have shown that lower activity of this biological process can lead to vulnerability in ischemia and nephrotoxicity
[176][180][181][182][183][184][185][186][184,188,189,190,191,192,193,194]. Likewise, balanced autophagy activity in the kidneys protects them from several renal insults
[187][188][189][190][195,196,197,198]. Interestingly, Klotho has been shown to be associated with autophagy levels
[191][192][193][194][199,200,201,202], although the exact molecular mechanism of this association is not yet completely understood.
Experiments with transgenic mice overexpressing Klotho have shown a positive correlation between higher autophagic flux and higher Klotho levels; at the same time, renal cells can become more vulnerable to oxidative stress when autophagy is suppressed. Furthermore, in cell culture experiments, autophagy inhibitors resulted in a decrease of Klotho’s induction of autophagy and cytoprotective effects against H
2O
2. It can inferred by these results that autophagy is one of the processes through which Klotho induces protection in renal cells, since its activity is upregulated by this enzyme
[195][203].
Further analysis has also shown that autophagy contributes to the maintenance of collagen balance in renal cells, according to experiments involving autophagy inducers and inhibitors to evaluate the expression of collagen type I (Col I) in OK cells. Likewise, transfection of OK cells with Klotho resulted in a decrease in Col I accumulation, both extracellular and intracellular. As this effect was abolished in part with the use of autophagy inhibitors, it has been suggested that this regulation of Col I promoted by Klotho might depend on autophagy flux
[195][203].
Moreover, there is evidence that due to the collagen degradation stimulated by autophagy
[176][196][197][184,204,205] and the induction of autophagy by Klotho, the amelioration of fibrosis in the kidneys promoted by Klotho might be associated with autophagy. Experiments in cell cultures with autophagy inhibitors have shown a decrease in Col I degradation by Klotho
[195][203].
Taken together, these results suggest that autophagy is one of the mechanisms through which Klotho protects the kidneys.