Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Romana Fato and Version 2 by Vivi Li.
Coenzyme Q (CoQ) is a key component of the respiratory chain of all eukaryotic cells. Its function is closely related to mitochondrial respiration, where it acts as an electron transporter. However, the cellular functions of coenzyme Q are multiple: it is present in all cell membranes, limiting the toxic effect of free radicals, it is a component of LDL, it is involved in the aging process, and its deficiency is linked to several diseases. Recently, it has been proposed that coenzyme Q contributes to suppressing ferroptosis, a type of iron-dependent programmed cell death characterized by lipid peroxidation.
- coenzyme Q10
- ubiquinone-10
- ubiquinol-10
- mitochondria
- OxPhos
- LDL
- statins
- age-related diseases
1. Introduction
Quinones are compounds widely present in nature, but their importance has increased considerably since they were recognized as fundamental components of the energy conservation systems present in all living organisms. The first quinone was discovered by Kofler, who was searching for compounds with Vitamin K activity in 1946 [1]. However, an accurate study of these molecules began only a decade later when ubiquinone(UQ), or Coenzyme Q (CoQ), was isolated in the laboratories of Richard Morton in Liverpool and David Green in Wisconsin. The presence of quinones had already been noted in biochemical studies on different phyla of living organisms. Crane and Lester’s studies on CoQ in electron transport led these authors to rediscover the role of Kofler’s quinone and to associate it with the electron transport mechanism in photosynthetic systems [2][3][4][5][6][7][2,3,4,5,6,7]
The chemiosmotic theory proposed by Mitchell in 1961 [8] introduced the revolutionary mechanism of transducing the energy associated with the protonmotive force into ATP synthesis, reinforcing the role of the CoQ in the mitochondrial electron transport chain. Indeed, the formation of trans-membrane ion gradients is the most ancient way to store energy in biological systems [9]. In fact, the presence of ATPases connected to redox enzymes was evidenced in LUCA (Last Universal Common Ancestor) and pre-LUCA organisms [10].
From the chemical point of view, ubiquinones show two important features: a redox active head (the benzoquinone ring) and a polyprenoid side chain varying in length between species. The side chain comprises ten isoprenoid subunits in humans (Ubiquinone-10), nine in mice (Ubiquinone-9), where ubiquinone-10 is also present as minor homolog, eight in E. coli, and six in S. cerevisiae. Although the electron and proton transfer activity is strictly associated with the benzoquinone ring, the polyprenoid tail is essential to restrict the molecule inside the lipophilic core of the biological membranes.
In addition to its bioenergetic role, CoQ has been associated with several important functions, such as the regulation of the cytosolic NAD+/NADH ratio and ascorbate reduction through the activity of the CoenzymeQ10 (CoQ10) dependent NADH-oxidase of the plasma membrane or PMOR, as it was originally termed by the researcher who first described it [11][12][13][11,12,13]. Then, after several years of research, it was discovered that Plasma Membrane Oxidase is a complex system, now indicated as Plasma Membrane Redox System (PMRS), involving different components; it serves as terminal oxidase for the cytosolic NADH and is responsible for the protein disulfide-thiol interchange activity on the outside of the plasma-membrane. Moreover, CoQ is a modulator of the mitochondrial transition permeability pore (mPTP) [14], is involved in gene regulation [15], and displays anti-inflammatory effects by influencing the expression of NFκ-β1-dependent genes [16][17][18][19][16,17,18,19]. In the reduced form, CoQ is a powerful antioxidant whose efficiency depends on its intramembranous distribution and the presence of many intracellular CoQ reducing enzymes.
Finally, the presence of CoQ is essential to maintain the integrity and functions of cell membranes [20][21][20,21].
2. Ubiquinone/Coenzyme Q Biosynthesis
Ubiquinone/Coenzyme Q is the only vitamin-like compound that can be completely endogenously synthetized. The biosynthetic process of CoQ in mammals is a very complicated pathway that consists of three distinct steps, located respectively in the cytosol for the isoprenoid side chain and for the synthesis of the aromatic precursor of the benzoquinone ring and in the mitochondrial matrix for CoQ assembly and benzoquinone ring modifications. The isoprenoid chain is synthesized through the mevalonate pathway, which leads to the farnesyl pyrophosphate (FPP), a common precursor of various metabolites: cholesterol, dolichols, isoprenylated proteins, and Coenzyme Q [22]. The regulatory step of the mevalonate pathway involves the 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA), the molecular target for statins. Noticeably, a well-known side-effect of statin therapy is statin-induced myopathy (SIM), associated with mitochondrial dysfunction and related to a reduced level of CoQ10, both in plasma and in muscle tissues [23][24][25][23,24,25]. In eukaryotes, the terminal steps in the biosynthesis of CoQ are thought to be rate limiting and occur in the mitochondrial matrix. In yeast, the mitochondrial assembly of CoQ requires at least 11 proteins encoded by COQ genes organized in a biosynthetic complex [26]. The human homologues of some COQ yeast genes were identified and, currently, mutations in COQ2 [27][28][27,28], PDSS1 [29], PDSS2 [30], COQ4 [31], COQ6 [32], ADCK3 [33][34][33,34], and COQ9 [35] have been described in patients with primary CoQ10 deficiency (OMIM # 607426). The assembly step in CoQ biosynthesis is catalyzed by the enzyme COQ2, a polyprenyl transferase that binds the polyprenoid chain to the six position of the p-hydroxybenzoic acid. Once assembled, the aromatic head of this CoQ precursor is modified to lead to the mature CoQ molecule. All the enzymes involved in these modifications are organized in a complex indicated as CoQ-synthome, which is localized on the matrix face of the inner mitochondrial membrane; this localization raises the problem of the access of the hydrophobic precursor to the modifying enzymes. The term ubiquinone refers to the ubiquitous presence of this molecule in all biological membranes and, besides the presence of a homolog of COQ2 in the endoplasmic reticulum (UBIAD1) [36], there is no evidence for an extramitochondrial CoQ biosynthesis. Given the extremely high hydrophobicity of this molecule, the problem of its distribution remains unsolved. Recently, in the group of Pagliarini, two proteins were identified, named Cqd1 and Cqd2, located in the inner mitochondrial membrane and involved in ubiquinone trafficking. Loss of Cqd1 favors the extramitochondrial distribution of CoQ leading to a CoQ deficient syndrome, even if the total amount of CoQ remains unchanged [37].2.1. Ubiquinone in Mitochondrial Electron Transfer Chains
Since its discovery, Ubiquinone or Coenzyme Q has been recognized as an obligate component of the mitochondrial respiratory chain. It is mainly localized in the inner mitochondrial membrane where it shuttles electrons from NADH-(Complex I) and Succinate-(Complex II) dehydrogenase to the bc1 complex (Complex III), then cytochrome c shuttles electrons to the cytochrome oxidase (Complex IV) directly responsible for O2 reduction. The free energy delivered by the electron transfer from the reduced coenzymes to the molecular oxygen drives the translocation of protons from the matrix site to the intermembrane space, generating the protonmotive force used by the mitochondrial ATPase to synthesize ATP.
As a component of the electron transfer chains, ubiquinone can exist in three different redox states. The fully oxidized form, ubiquinone (CoQ), can be reduced by the two-step gain of two electrons and two protons to give the fully reduced form (ubiquinol form or CoQH2), passing through a radical form (the semiquinone form or CoQH) (Figure 1).
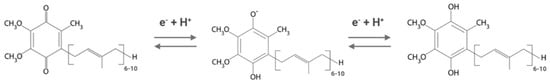
Figure 1. Different redox states of Coenzyme Q. From left to right: oxidized form (ubiquinone), semi-reduced form (semiquinone), and fully reduced form (ubiquinol). Depending on the organism, the isoprenoid chain is composed of 6 to 10 units.
While the bioenergetic role of CoQ is clear, the structural organization of the respiratory chain and the mechanism by which electrons are transferred from reduced coenzymes to molecular oxygen are still a matter of debate. Following the observation that the concentration of ubiquinone was more than the concentration of the prosthetic groups in the redox enzymes, Green and Tzagoloff first introduced the idea of CoQ as a mobile electron carrier [38]. This hypothesis was supported by the pioneering work of Kroeger and Klingenberg [39], who derived a simple equation to describe the kinetic behavior of ubiquinone, concluding that all the quinones present in the inner mitochondrial membrane behave like a common pool and the rate of electron transfer depends only on the rate of input and output of electrons to the quinone’s pool. For the Random Diffusion Model of Electron Transfer chain [40], the redox enzymes are randomly distributed into the lipid bilayer and electron transfer is ensured by free diffusion of ubiquinone and cytochrome c. Nevertheless, about 15 years later, Schagger and Pfeiffer reintroduced the hypothesis of a solid-state organization of the respiratory chain based on structural evidence by blue native electrophoresis [41][42][41,42]. Subsequently, several pieces of evidence favoring alternatively solid-state organization or random distribution appeared in literature, leaving the problem unsolved.
Complex II is not recognized as a component of this supramolecular organization. The original hypothesis of the common quinone pool, introduced by Kroeger and Klingenberg [39] in 1971, was replaced by assuming the presence of two different quinone pools: one acting for the oxidation of NADH and the second for the oxidation of succinate and for the oxidation of substrates of other dehydrogenases that use ubiquinone as an electron acceptor [43][44][43,44]. However, no quinones were found permanently bound to respiratory enzymes, leading to speculation of a dynamic equilibrium between quinone pools.
Although the role of UQ is to transfer electrons from dehydrogenases to the bc1 Complex, in the presence of a high protonmotive force and a high level of reduced ubiquinone (CoQH2) it is possible to observe a back-flow of electrons from ubiquinol to NAD+ involving Complex I. This Reverse Electron Transfer (RET) has been recognized since the second half of the 1960s [45], but it was considered an in vitro phenomenon observed in isolated mitochondria without physiological relevance [46][47][48][46,47,48]. RET was associated with an increased Reactive Oxygen Secies (ROS) production from Complex I, especially in conditions of ischemia-reperfusion injury [49][50][49,50]. During ischemia, the low level of oxygen would be responsible for the complete reduction in the respiratory chain components, in particular, the high ratio CoQH2/CoQ would result in a concomitant increase in ROS production. Recently, the role of the redox state of Coenzyme Q was reappraised, confirming the crucial function of the ratio of CoQH2/CoQ as an endogenous sensor for the mitochondrial function [51][52][51,52]. Low oxygen levels or impairment of Complex III/Complex IV would result in an excess of CoQ reduced form, due to an unbalancing between the quinone reducing enzymes activity and the quinol oxidizing enzymes. Excess reduced quinone would induce reverse electron transfer by stimulating ROS production from Complex I, which in turn would cause a partial degradation of mitochondrial Complex I itself. The purpose of this vicious cycle is to avoid excessive ROS production that would be deleterious to cell survival.
Besides Complex I and II, several dehydrogenases, mostly FAD-dependent, can fuel electrons to the ubiquinone pool, for example, ETF-dehydrogenase, involved in fatty acid beta-oxidation, or dihydroorotate dehydrogenase, which is essential for the de novo synthesis of pyrimidine nucleotide. In addition to the above, glycerol-3-phosphate dehydrogenase, which connects glycolysis and fatty acid metabolism with the oxidative phosphorylation [53], and proline and choline dehydrogenases, which use CoQ as electron acceptor, are included [54][55][54,55] (Figure 2). Finally, a quinone dependent D-lactate dehydrogenase has been recognized to be over-expressed in cancer cells [56], where it contributes to the detoxification of the methylglyoxal, a toxic metabolite derived by aberrant glucose catabolism and associated with cancer progression and Alzheimer’s disease [57][58][57,58]. A metabolic switch from glucose to fatty acid metabolism affects electron entry into the respiratory chain, favoring the activity of flavin-dependent dehydrogenases over NADH-dependent dehydrogenases. This will lead to an excess of FADH2 oxidation with a concomitant increase in the reduced state of the CoQ pool, which is considered responsible for the reverse electron transfer and the increased ROS production from mitochondrial Complex I. Szibor M et al. demonstrated that the expression of an alternative oxidase (AOX) could oxidize the quinone pool, releasing the excess of reduced quinone and inducing a shift from RET to FET (Forward Electron Transfer) [59].
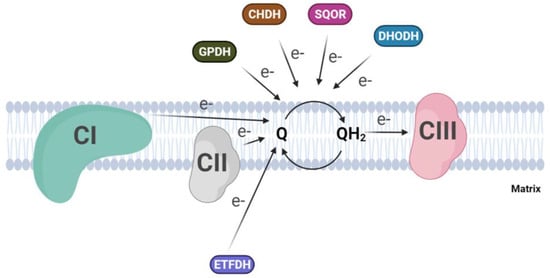
Figure 2. Convergent electron flow to the Coenzyme Q pool in the inner mitochondrial membrane. (CI, Complex I; CII, Complex II; CIII, Complex III; GPDH, Glycerol-3-phosphate dehydrogenase; CHDH, choline dehydrogenase; SQOR, sulphide:quinone oxidoreductase; DHODH dihydroorotate dehydrogenase).
2.2. CoQ10 in Cell Membranes
In non-mitochondrial biological membranes, CoQ continuously switches between reduced and oxidized forms through the action of enzymes with CoQ reductase activity [60]. These enzymes encompass NAD(P)H dehydrogenases, which are part of the plasma membrane redox system (PMRS). Among these reductases, CytB5R3 [61] and NQO1 [62] are the most important enzymes which maintain ubiquinol levels in cell membranes [63]. The PMRS becomes essential for the maintenance of bioenergetics when the mitochondrial activity is reduced, such as in aging. PMRS upregulation has been demonstrated in mtDNA-deficient cell lines with cytosolic NADH accumulation. Through NADH re-oxidation, PMRS allows cells to maintain the correct NAD+/NADH ratio required to sustain glycolytic flux [21].
In a recent paper [64], reswearchers demonstrated that in a human glioma cell line (T67) treated with 4-NB, a competitive inhibitor of the COQ2 enzyme, the endogenous level of CoQ10 decreased by 50%. This CoQ depletion, in addition to a decrease in the mitochondrial respiratory chain activity, induces a decrease also in the PMRS activity, confirming the crucial role of CoQ in maintaining this extra-mitochondrial electron transfer activity. Researchers'Our results showed that, although the cells try to counteract the mitochondrial dysfunction upregulating the PMRS enzymes, the CoQ10 depletion induced a strong decrease in the efficiency of the plasma membrane redox system. Exogenous CoQ supplementation induces a strong increase in the cellular ubiquinone content, restores the PMRS efficiency, and increases the mitochondrial oxygen consumption rates. Although CoQ supplementation increased the PMRS efficiency over control levels, it was not able to completely restore the mitochondrial oxygen consumption rate, suggesting the existence of a rate-limiting step in the intracellular distribution for a so strongly hydrophobic molecule.
Besides its role in the redox state regulation, together with PMRS activity, CoQ plays an important role as a lipid-soluble antioxidant, preventing lipid peroxidation in biological membranes. It represents the only endogenously synthesized lipid-soluble antioxidant capable of protecting lipids, proteins, and DNA from oxidative damage [16]. Unlike other antioxidants, ubiquinol can inhibit both the initiation and propagation of oxidative damage, preventing the formation of lipid peroxyl radicals (LOO•); it reacts with preferryl radical and radicals generating ubisemiquinone and a non-radical lipid hydroperoxide [22][65][22,65]. Moreover, UQH2 effectively regenerates vitamin E from the α-tocopheroxyl radical, which additionally contributes to slow the propagation step of lipid peroxidation.
The exceptionally high antioxidant efficiency of CoQ is due to its intra-membrane localization, its general and abundant distribution, and its effective reduction/reactivation by several cellular systems that catalyze its reduction to the active form [16]. In mitochondria, the antioxidant reduced form of CoQ is regenerated directly by the respiratory chain [19]. In addition to the mitochondrial respiratory chain, the PMRS also exerts the same function, generating ubiquinol by transferring two electrons in a two single-step mechanism (CytB5R3) [61] or by direct two-electrons quinone reduction (NQO1) [62]; this last mechanism avoids the formation of a semiquinone intermediate (Figure 3).
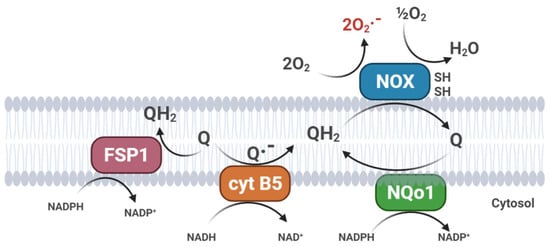
Figure 3. Plasma membrane redox systems (PMRS). (FSP1, Ferroptosis Suppressor Protein 1; cyt B5, cytochrome b5 reductase; NQo1, NAD(P)H: quinone oxidoreductase; NOX, plasma membrane NADH oxidase).
Mitochondria also play a role in ferroptosis, a unique mode of cell death, driven by iron-dependent phospholipid peroxidation. Ferroptosis is regulated by multiple cellular metabolic pathways; the cyst(e)ine–GSH–GPX4 axis is considered the main system opposing ferroptosis in mammals [66]. Recently, the CoQ oxidoreductase ferroptosis suppressor protein 1 (FSP1) has been shown to inhibit ferroptosis together with the glutathione peroxidase 4 (GPX4) [67]. Flavoprotein AIFM2 (apoptosis-inducing factor mitochondria-associated 2), previously characterized as a pro-apoptotic and P53-responsive protein associated with the mitochondrial outer membrane, is now considered as a novel ferroptosis modulator and renamed FSP1 (ferroptosis suppressor protein 1). Myristoylated FSP1 is associated with several cell membrane structures, including the Golgi apparatus, perinuclear structures, and the plasma membrane, where it serves as an NADPH-dependent oxidoreductase to mediate the reduction of ubiquinone and trap lipid peroxyl radicals, thus suppressing lipid peroxidation [66] (Figure 3). In a recent paper, rwesearchers demonstrated that, in cultured cells, exogenous CoQ supplementation can protect membrane lipids from peroxidation and increase the resistance of cells to ferroptotic stimuli, such as treatment with erastin or RLS3, an inhibitor of GPX4 [68]. Interestingly, ferroptosis is associated with several neurodegenerative disorders (Parkinson’s and Alzheimer’s diseases), chronic inflammation, frailty, and cancer; therefore, CoQ10 supplementation could be beneficial to counteract the age-related progression of these diseases [69].
2.3. CoQ as a Component in Low Density Lipoproteins (LDL)
Besides its cellular involvement in bioenergetic functions, CoQ is also a component of LDL; this suggests its potential role in atherosclerotic prevention [70][71][70,71]. The mode of action in protecting the LDL from oxidation is similar to the one described for cell membranes, as previously discussed, together with α tocopherol [71]. CoQ and α tocopherol protect from cellular damage in proportion to the amount of their content in the LDL membrane, suggesting that supplementation with these molecules could overcome LDL oxidation.
Interestingly, HepG2 cells can reduce extracellular CoQ10 to CoQ10H2 using reductase present on the outer surface of cells, providing reduced levels of CoQ10 in plasma [72]. Moreover, CoQ10, vitamin E, and dihydro thioctic acid can cooperatively act to prevent LDL oxidation [73].
The double-edged knife of cardiovascular risk prevention is well described by the negative action of pharmacological prevention with statins on physiological CoQ10 levels. Statins block endogenous production of cholesterol in the liver by inhibiting 3-hydroxy-3-methylglutaryl-Coenzyme A (HMG-CoA) reductase [74], affecting CoQ synthesis and thus accelerating CoQ depletion in elderly people.
The negative effects of long-term utilization of statin, in the primary and secondary intervention of lowering cardiovascular risk, are evident mostly in muscle and liver in selected groups of elderly people. The reason for such side effects could reside in genetic predisposition. The gene SLCO1B1 (solute carrier organic anion transporter family member 1B1) codes for an organic anion-transporting polypeptide that is involved in the regulation of the absorption of statins. A common variation in this gene was found to significantly increase the risk of myopathy [75].
2.4. Aging and Longevity: Role of CoQ10 Homeostasis
Exercise and oxygen consumption through the respiratory chain usually produce free radicals in the form of ROS (Reactive Oxygen Species), which participate in regular hormetic response [76]. The hormetic response consists of, among other things, a major production of antioxidant enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx). Furthermore, to protect macromolecules from oxidative damage, cells contain antioxidant molecules such as: ascorbic acid, alpha-tocopherol, glutathione, and coenzyme Q10 (CoQ10), which are used by the antioxidant enzymes and other systems, such as cytochrome b5 reductase (CytB5R3) or NAD(P)H quinone dehydrogenase 1 (NQO1). Currently, it has been established that aging is associated with an accumulation of malfunctioning mitochondria at the cellular and tissue level [77][78][79][77,78,79]. Because of its central role in mitochondrial physiology, a decrease in CoQ10 levels could contribute to accelerating the dysfunction of mitochondrial activity associated with aging and related diseases [80]. CoQ10 is also a potent antioxidant at the level of the cellular plasma membrane and in plasma lipoproteins. The reduced form of CoQ10, ubiquinol (UQH2), prevents both the initiation and propagation of lipid peroxidation in cell membranes [81] and plasma lipoproteins. CoQ10 also regulates gene expression, especially of genes involved in the chronic inflammatory response, and exhibits anti-inflammatory properties [15][82][83][15,82,83]. Therefore, it seems clear that antioxidant integration is necessary to counteract the negative effects of age-related ROS accumulation. The central physiological role of CoQ10 points to it as the best antioxidant to supplement. CoQ10 is also involved in several cellular functions that should be studied in detail to target an effective supplementation intervention for aging and age-related disorders.
2.5. CoQ in Human Diseases
In human disease, CoQ10 deficiency has been described both as a primary and secondary defect and has been involved in the pathogenesis of different conditions (Figure 4). Primary CoQ10 deficiencies are rare autosomal recessive disorders that can affect many organs and tissues, especially the brain, muscles, and kidneys, involving a shortage (deficiency) of CoQ10 due to mutations in the genes involved in the biosynthesis of CoQ10. Secondary CoQ10 deficiencies are caused by defects in mitochondrial and non-mitochondrial processes associated with a secondary reduction in CoQ10 in cells or tissues.
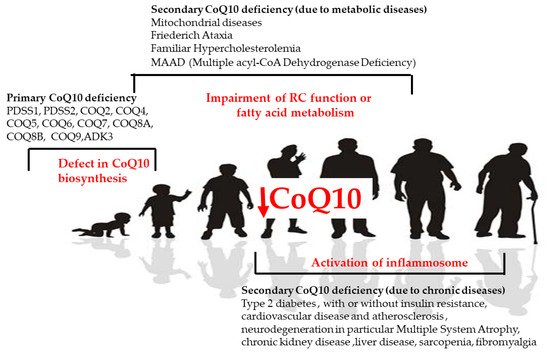
Figure 4.
CoQ in human diseases. Causes of primary and secondary deficiency of CoQ10 in human disease.
Different clinical phenotypes can be strictly related to CoQ10 deficiency at a specific tissue level. Primary CoQ10 deficiencies are characterized by five major phenotypes due to mutations in any of the genes involved in CoQ10 biosynthesis, namely encephalomyopathy, cerebellar ataxia, infantile multisystemic form, isolated myopathy, and nephropathy [84]. Up to now, mutations in PDSS1, PDSS2, COQ2, COQ4, COQ5, COQ6, COQ7, COQ8A, COQ8B, and COQ9 genes have been found to cause human CoQ deficiency disease, but only about 280 patients from 180 families have been reported in the literature.
The extreme variability in clinical and phenotypic presentation, such as the age of onset, severity of disease, and organs involved, make the existence of a unique common pathway for all the forms unlikely. Thus, recently, the presence of a possible unknown function of CoQ10 genes not strictly related to oxidative phosphorylation (OXPHOS) function is starting to be postulated. A therapy with CoQ10 supplementation is actually present in the clinical practice. Although this compound is used and could give good therapeutic prospects, no clinical trials or long-term phase III studies have yet been performed to truly document and prove the efficacy of this compound in either primary or secondary CoQ10 deficiency. In the primary deficit of CoQ10 such clinical trials might be difficult to implement, first because of the very low number of patients, and also because of the severity of the diseases, the limited data on the natural history of these diseases, and, ultimately, the few outcome measures available.
Quinzii et al. described an accumulation of hydrogen sulfide (H2S) in primary CoQ deficiencies, emphasizing one specific role of CoQ10 with the participation of the sulfide oxidation pathway via sulfide:quinone oxidoreductase (SQOR) [85]. The close relationship between H2S and CoQ levels can explain these findings in primary CoQ disorders, in which H2S is formed by desulfuration of homocysteine and cysteine via Cystathionine-β synthase (CBS) and cystathionine γ lyase (CSE). High levels of H2S seem to have deleterious effects as COX inhibitors, while at the physiological level can be utilized by the Krebs cycle as a substrate. Unbalanced homocysteine levels, in turn, can decrease H2S levels [86], as observed in hyperhomocysteinemia related to cardiovascular diseases. Regulation of H2S levels can thus provide beneficial effects both in primary and secondary CoQ10 deficiencies, becoming a key point in therapeutic approaches.
CoQ10 deficiency is much more common in association with other diseases, either related to a specific genetic condition not directly involved in CoQ10 biosynthesis or in nongenetic disorders as a side effect. Secondary CoQ10 deficiency is often present in mitochondrial disease in which CoQ10 biosynthesis genes are not directly involved, but with a clinical presentation similar to the ones described in many mitochondrial diseases, such as cardiomyopathy or myopathy, exercise intolerance, or brain diseases and epilepsy. In these diseases, the administration of a high dose of CoQ10 is a common therapy even if a clear efficacy has not yet been proved. Moreover, a deficit in CoQ10 has been identified in some genetic disorders linked to lipid metabolism, such as Familial Hypercholesterolemia and MAAD (Multiple acyl-CoA Dehydrogenase Deficiency) [87]. In both these conditions, the administration of CoQ10 lead to a reduction in the atherosclerosis and to a reduction in muscle pain and fatigue. More recently, a role in CoQ10 in inflammation and in autoimmune diseases has been postulated, with it being proven that CoQ10 supplementation reduced the levels of circulating inflammatory markers. This CoQ10 anti-inflammatory potential might be related to the downregulation of genes regulated by nuclear factor-κβ (NF-κβ), which is known to be activated by reactive oxygen species [88].
The role of CoQ10 has been described in fibromyalgia, a chronic pain syndrome whose pathophysiological mechanisms have not yet been identified, and a decrease in mitochondrial mass and CoQ10 levels, as well as an overproduction of ROS, was detected in blood mononuclear cells from patients affected by fibromyalgia [89]. Moreover, the pain in fibromyalgia could also be mediated by the activation of the inflammasome by CoQ10 [90]. The role of CoQ10 in inflammation could be in part responsible for some secondary mitochondrial dysfunction, and CoQ depletion is also present in different chronic age-related disorders such as type 2 diabetes [91], with or without insulin resistance, cardiovascular disease and atherosclerosis [92], neurodegeneration, in particular Multiple System Atrophy [93], chronic kidney disease [94], liver disease [95], and sarcopenia [96].