Transcription factor NF-κB has been extensively studied for its varied roles in cancer since its initial characterization as a potent retroviral oncogene several decades ago. It is now clear that NF-κB plays a major role in a large variety of human cancers, including especially ones of immune cell origin. NF-κB is generally constitutively or aberrantly activated in human cancers where it is involved.
- NF-kappaB
- cancer
- therapy
- immunity
- signal transduction
1. Introduction
Eukaryotic transcription factor NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) has been the subject of intense study over the past several decades for its role in a variety of normal and pathological processes[1]. Indeed, there are currently approximately 100,000 publications with information related to NF-κB. This entry features a historical and conceptual overview of NF-κB signaling and human cancer, as well as therapeutic implications. As such, it is meant to serve as a broad introduction to more specific articles on NF-κB and cancer.
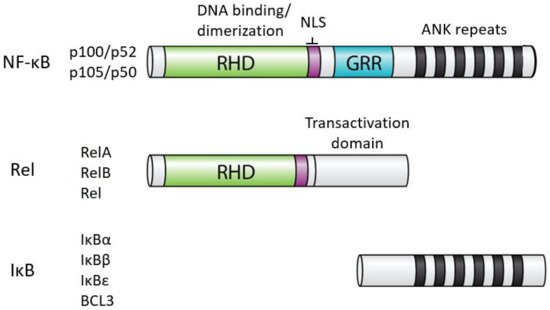
In humans, NF-κB comprises five related proteins (p52, p50, RelA, RelB, Rel) that bind DNA response elements as dimers to control gene expression. The NF-κB proteins are related by an approximately 300 amino acid domain called the Rel Homology Domain (RHD), which contains sequences necessary for DNA binding, dimerization, and nuclear localization (Figure 1). The five human NF-κB proteins can form essentially all combinations of homodimers and heterodimers, and the composition of the dimer, at least in part, dictates the precise DNA sequence they can bind.
In almost all normal cell types, NF-κB is located in the cytoplasm in an inactive state. When activated (often by extracellular signals), NF-κB moves to the nucleus, and binds to DNA to modulate the transcription of specific target genes for a given biological response. The activity of all NF-κB dimers is modulated by interaction in the cytoplasm with a family of NF-κB inhibitor proteins called IκBs[2]. All IκB proteins consist of a series of 5–8 ANK repeats, which are protein interaction domains that interact with the RHD sequences and block the ability of NF-κB dimers to bind to DNA and translocate to the nucleus. There are several independent IκB proteins (e.g., IκBα, -β, -ε, BCL3) which have different cell type-specific expression and have different affinities for the different NF-κB dimers. In addition, the C-terminal sequences of p100 and p105 contain ANK repeat IκB sequences that interact intramolecularly with the RHD sequences to block their activity. The general structures of NF-κB and IκB proteins are shown in Figure 1.
The first evidence for a role of NF-κB in cancer came from the characterization of the v-rel oncogene of the avian Rev-T retrovirus[3]. Indeed, Rev-T is an extremely potent oncogenic agent and causes a rapidly lethal lymphoma when injected into young chickens. Moreover, substitution of the human REL proto-oncogene for v-rel in Rev-T is also highly oncogenic in chicken lymphoid cells, both in vivo and in vitro.
In spite of its potency as a single-agent oncogene in avian systems, NF-κB has not emerged as an efficacious mammalian oncogene in the manner of RAS, MYC, or SRC. Namely, no NF-κB protein has been shown to have rapid and strong oncogenic activity in any mammalian cell line or transgenic mouse model. Nevertheless, constitutively active NF-κB has been found in over 40 cancer types (Table 1), and this activity has been implicated in a variety of standard cancer-associated biological processes, including cell survival, cell proliferation, metastasis, inflammation, angiogenesis, immune cell inhibition, growth factor activity, and stromal cell effects[4]. Relevant NF-κB target genes for many of these biological situations have been identified.
Table 1. Cancers that have been reported to have high NF-κB activity.
| Hematological Malignancies | Solid Tumors |
|---|---|
| Hodgkin lymphoma | Breast |
| Acute lymphoblastic leukemia | Cervical |
| Acute myelogenous leukemia | Ovarian |
| Acute T cell leukemia | Vulvar |
| Acute nonlymphocytic leukemia | Uterine (endometrial) |
| Chronic lymphocytic leukemia | Prostate |
| Chronic myelogenous leukemia | Testicular |
| Burkitt lymphoma (EBV) | Penile |
| Mantle cell lymphoma | Kidney |
| Myelodysplastic syndrome | Bladder |
| Multiple myeloma | Lung |
| Diffuse large B-cell lymphoma | Mesothelioma |
| MALT lymphoma | Esophageal |
| Mantle cell lymphoma | Laryngeal |
| Marginal zone lymphoma | Liver |
| Waldenstrom’s macroglobulinemia | Pancreatic |
| Stomach | |
| Colon | |
| Thyroid | |
| Parathyroid | |
| Melanoma | |
| Squamous cell carcinoma | |
| Head and neck | |
| Cylindromatosis | |
| Trichoepithelioma | |
| Hilar cholangiocarcinoma | |
| Oral carcinoma | |
| Tongue | |
| Astrocytoma | |
| GIST |
For details, see http://www.bu.edu/nf-kb/physiological-mediators/diseases/.
2. Cancer Cell-Induced Activation of NF-κB
Unlike what is found in most normal cells, NF-κB is constitutively located in the nucleus in many cancer cells. Constitutive activation of NF-κB can occur by several mechanisms and in a large number of different cancers.
NF-κB activity can be activated by cancer-associated gene mutation. These mutations can occur in both core NF-κB pathway proteins (e.g., as ctivating mutations or amplifications of REL or p52/p100 or deletions of IκB inhibitors), as well as in NF-κB upstream regulators. Such mutations are perhaps best characterized in hematological malignancies, including prominently B-cell and T-cell leukemias and lymphomas[5]. Nevertheless, cancer genome sequencing projects are now uncovering NF-κB pathway mutations in many other cancer types.
The most highly studied malignancies with NF-κB mutations are the relatively common malignancies diffuse large B-cell lymphoma (DLBCL)[6] and multiple myeloma[7]. Indeed, mutation-based activation of NF-κB defines a molecular subtype of DLBCL, which has a poorer clinical outcome in response to standard chemotherapy. The NF-κB-dependent subtype of DLBCL has high expression of several direct NF-κB target genes, which are generally involved in sustained cell proliferation and prolonged cell survival. Activating mutations of NF-κB occur in about 40% of DLBCL and most commonly occur at multiple steps in the B-cell receptor (BCR)-to-NF-κB signaling pathway[8]. As such, some gain-of-function mutations generate chronic activators of NF-κB, whereas other loss-of-function mutations inactivate negative regulators of NF-κB[8]. Similarly, multiple pathway activators can lead to constitutive NF-κB pathway activation in multiple myeloma (MM) in about 20-40% of cases[9]. Moreover, NF-κB pathway inhibition can block the proliferation and survival of DLBCL and MM cells that have chronic activation of NF-κB[10].
Nevertheless, it is important to note that mutation-based activation of NF-κB is simply one oncogenic driver pathway in most B-cell malignancies. That is, there are a variety of other cooperating pathway mutations, e.g., in proteins such as NOTCH, BCL6, IRF4, p300/CBP, that cluster with NF-κB pathway[11]. And, in some cases, these alterations are required to keep NF-κB activity in an optimal range for the malignant process.
Probably the most common type of mutation that leads to enhanced NF-κB signaling occurs in genes encoding upstream or downstream proteins in specific pathways. In DLBCL and MM, this can include mutations in cell-surface receptors, in pathway adaptors and pathway modulators[5]. In short, DLBCL and MM are essentially genetic experiments that select for tumor cells that have mutations that drive sustained activation of NF-κB, which is required for malignant B-cell proliferation and survival. Similar types of activating mutations are also found in many of the cancers listed in Table 1.
3. NF-κB as a Tumor Suppressor
In a limited number of cases, NF-κB has been reported to have tumor suppressor activity[12]. However, this activity has generally been demonstrated in mouse or cell-based models. For example, downregulation of NF-κB by overexpression of an IκB protein led to skin epidermal hyperplasia in mice. However, a direct oncogenic effect of reduced NF-κB activity is not yet clear, and tumor suppressor activity of NF-κB has not been well-documented in human cancers[13].
4. Targeting of NF-κB for Cancer Therapy
Based on the pervasive involvement of NF-κB in oncogenesis, it is not surprising that NF-κB signaling would be considered as a target for human cancer therapy. Indeed, there are well over 1000 inhibitors that can block NF-κB signaling [14], and inhibition of NF-κB can block cancer cell proliferation or survival in a number of animal models. Nevertheless, inhibitors of NF-κB signaling have not had a dramatic impact on general cancer therapy in humans, in part because of the liver toxicity of many NF-κB inhibitors and the rapid development of parallel pathway resistance. Still, proteasome inhibitors (which block NF-κB activation) have been quite effective for the treatment of multiple myeloma, and they appear to act, at least in part, due to their inhibitory effects on NF-κB signaling[10]. Similarly, Bruton’s tyrosine kinase (BTK) inhibitors have been useful in the treatment of NF-κB-positive DLBCL[15]. Finally, whether inhibition of NF-κB might promote cancer in some cases by affecting tumor-suppressing activity of NF-κB or by having a detrimental effect on patient antitumor T cell immunity (natural or induced by immunotherapy) is not clear.
It is likely that dysregulation of NF-κB will be discovered to be associated with many more cancers, especially as additional whole genome sequence data of cancers is accumulated. Nevertheless, it is unlikely that sustained, systemic, and complete inhibition of upstream NF-κB pathways will be a useful primary strategy for cancer. That being said, agents and doses that reduce (but do not eliminate) cancer-associated NF-κB activity or inflammation may well be useful. On compelling need is for therapies that can distinguish and target oncogenic NF-κB activity versus normal NF-κB activity, i.e., to avoid unwanted side effects on normal tissues,
Overall, it is almost certain that research on pathological NF-κB activity will continue to occupy an important place in cancer research. Modern genome modification methods and/or new therapeutic strategies to target cancer-associated NF-κB activity may prove useful in certain clinical settings.
References
- Gilmore, TD.; Introduction to NF-κB: Pathways, players, perspectives. Oncogene 2006, 25, 6684-6690.
- Hayden, MS, Ghosh, S.; Shared principles of NF-κB signaling. Cell 2008, 132, 344-362.
- Gilmore, TD; Multiple mutations contribute to the oncogenicity of the retroviral oncoprotein v-Rel. Oncogene 1999, 18, 6925-6937.
- Xia Y, Shen S, Verma IM; NF-κB, an active player in human cancers. Cancer Immunology Research 2014, 2, 823-830.
- Lim K-H, Yang Y, Staudt LM; Pathogenetic importance and therapeutic implications of NF-κB in lymphoid malignancies. Immunological Reviews 2012, 246, 359-378.
- Staudt LM; Oncogenic activation of NF-κB. Cold Spring Harbor Perspectives in Biology 2010, 2, a000109.
- Matthews GM, de Matos Simoes R, Dhimolea E, Sheffer M, Gandolfi S, Dashevsky O, Sorrell JD, Mitsiades CS; NF-κB dysregulation in multiple myeloma. Seminars in Cancer Biology 2016, 39, 68-76.
- Young RM, Phelan JD, Wilson WH, Staudt LM; Pathogenic B-cell receptor signaling in lymphoid malignancies: New insights to improve treatment. Immunological Reviews 2019, 291, 190-213.
- Roy P, Aditya Sarkar U, Basak S; The NF-κB activating pathways in multiple myeloma. Biomedicines 2018, 6, 59.
- Vrábel D, Pour L, Ševcková S; . The impact of NF-κB signaling on pathogenesis and current treatment strategies in multiple myeloma.. Blood Reviews 2019, 34, 56-66.
- Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al.; Genetics and pathogenesis of diffuse large B-cell lymphoma. New England Journal of Medicine 2018, 378, 1396-1407.
- Perkins, ND; The diverse and complex roles of NF-κB subunits in cancer. Nature Reviews Cancer 2012, 12, 121-132.
- Chen F, Castronova V; Nuclear factor-κB, an unappreciated tumor suppressor. Cancer Research 2007, 67, 11093-11098.
- Gilmore TD, Garbati MR; Inhibition of NF-κB signaling as a strategy in disease therapy. Current Topics in Microbiology and Immunology 2011, 349, 245-263.
- Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, Shaffer AL, Ceribelli M, Wang JQ, Schmitz R, et al.; A multiprotein supercomplex controlling signaling in lymphoma. Nature 2018, 560, 387-391.