The heart is a metabolic omnivore that combusts a considerable amount of energy substrates, mainly long-chain fatty acids (FAs) and others such as glucose, lactate, ketone bodies, and amino acids. There is emerging evidence that muscle-type continuous capillaries comprise the rate-limiting barrier that regulates FA uptake into cardiomyocytes. The transport of FAs across the capillary endothelium is composed of three major steps—the lipolysis of triglyceride on the luminal side of the endothelium, FA uptake by the plasma membrane, and intracellular FA transport by cytosolic proteins. In the heart, impaired trans-endothelial FA (TEFA) transport causes reduced FA uptake, with a compensatory increase in glucose use. In most cases, mice with reduced FA uptake exhibit preserved cardiac function under unstressed conditions. When the workload is increased, however, the total energy supply relative to its demand (estimated with pool size in the tricarboxylic acid (TCA) cycle) is significantly diminished, resulting in contractile dysfunction. The supplementation of alternative fuels, such as medium-chain FAs and ketone bodies, at least partially restores contractile dysfunction, indicating that energy insufficiency due to reduced FA supply is the predominant cause of cardiac dysfunction.
- cardiac metabolism
- fatty acid
- capillary endothelium
- trans-endothelial fatty acid transport
- contractile function
1. Mechanisms of FA Uptake by the Heart
1.1. Source of Long-Chain Fatty Acids
1.1. Source of Long-Chain Fatty Acids
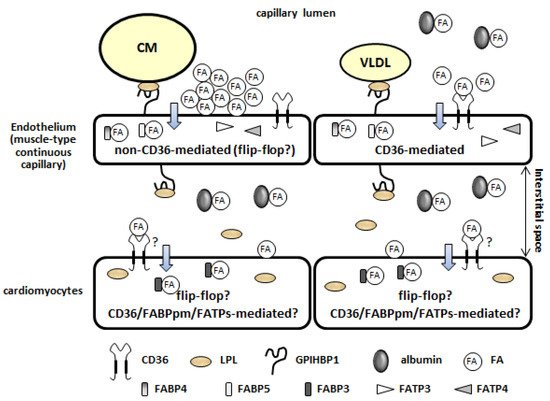
1.2. Lipolysis of TG Contained in TG-Rich Lipoproteins on the Luminal Side of the Capillary Endothelium
LPL is an essential enzyme that hydrolyses the TG contained in TGRLPs [1][2][3][4]. Importantly, LPL is predominantly produced in cardiomyocytes and is transferred to the luminal side of the endothelium, where the enzyme functions (1.2. Lipolysis of TG Contained in TG-Rich Lipoproteins on the Luminal Side of the Capillary Endothelium
1.3. Fatty Acid Uptake by the Plasma Membrane of the Capillary Endothelium (Non-CD36-Mediated and CD36-Mediated Pathways)
There are two distinct pathways of FA uptake by the capillary endothelium [9][10]—a high-capacity non-saturable pathway (1.4. Intracellular Fatty Acid Transport through the Capillary Endothelium
Following FA uptake via the plasma membrane, intracellular FA transport is performed by cytosolic proteins. Fatty acid-binding proteins 4 and 5 (FABP4/5), abundantly expressed in the capillary endothelium in the heart, are potential candidates for transport (1.4. Intracellular Fatty Acid Transport through the Capillary Endothelium
1.5. Fatty Acid Uptake by Cardiomyocytes
Following TEFA transport (lipolysis, FA uptake by the plasma membrane, and intracellular FA transport), FAs are bound by albumin (300 μM) in the interstitial space of the heart (1.5. Fatty Acid Uptake by Cardiomyocytes
2. Molecular Mechanisms Underlying the Induction of Genes Associated with Trans-Endothelial Fatty Acid Transport
Recent studies have revealed that the expression of genes associated with TEFA transport is regulated by several ligands, receptors, and transcription factors (Table 1) [21][22][23][24][6,7,8,9]. It is likely that these systems can be roughly divided into two groups according to their target genes. One includes the peroxisome proliferator-activated receptor γ (PPARγ), mesodermal homeobox-2/transcription factor 15 (Meox2/Tcf15), Notch signaling, and the apelin/apelin receptor (APLNR), and it mainly controls the expression of CD36, FABPs, and GPIHBP1. The other is a group that includes the VEGF-B/VEGF receptor (VEGFR), angiopoietin-like 2 (ANGPTL2), and 3-hydroxyisobutyrate (3-HIB), and it regulates the expression/function of FATP3/4 (Table 1). Although impairments of the systems influence both local and systemic metabolism, cardiac metabolism seems to only be affected by PPARγ, Meox2/Tcf15, Notch signaling, and VEGF-B/VEGFR (Table 1) [21][22][23][24][6,7,8,9]. The trans-endothelial transport of other substrates and molecules (e.g., lipoproteins, lipoprotein lipase, glucose, and insulin) and endothelium-derived metabolic regulators (e.g., nitric oxide, extracellular matrix proteins, hormones, growth factors, and enzymes) is described elsewhere [21][22][24][6,7,9].
2.2. Mesodermal Homeobox-2/Transcription Factor 15
Meox2 is a homeobox gene that is expressed in the microvascular endothelium in the heart [28]. Meox2 forms a heterodimer with a basic helix–loop–helix Tcf15, which is highly expressed in the capillary endothelium. The Meox2/Tcf15 heterodimer drives the endothelial expression of genes associated with FA metabolism, including PPARγ, CD36, FABP4/5, LPL, and GPIHBP1, to facilitate FA uptake and transport across the capillary endothelium [28]. Importantly, the haplodeficiency of Meox2/Tcf15 in mice was found to cause reduced FA uptake with compensatory glucose use in the heart (2.1. Peroxisome Proliferator-Activated Receptor γ
2.2. Mesodermal Homeobox-2/Transcription Factor 15
2.3. Notch Signaling
Notch signaling is not only a master regulator of angiogenesis but also a regulator of TEFA transport. The inhibition of endothelial Notch signaling in the adult heart leads to reduced FA transport, resulting in heart failure and hypertrophy [29][30]. The activation of Notch signaling facilitates FA transport by inducing CD36, FABP4/5, and lipase G endothelial type (LIPG) and by suppressing angiopoietin-like 4 (ANGPTL4), a well-characterized inhibitor of LPL [29][30].2.3. Notch Signaling
| Target Genes | Deficient Site | Inducible Knockout | VLDL-TG Uptake | FA Uptake | Glucose Uptake | Glut1/4 | Ketonein Serum | Contractile Performance In Vivo Estimated by Echocardiography | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LPL (functions at luminal side of capillary) | cardiomyocyte | ↓ | ↑ | ↑ | ↑ | ↓ aged | [37] | [44] | |||||
| cardiomyocyte | ⚪ | ↓ | [38] | [45] | |||||||||
| CD36 | whole | ↓ | ↑ | ↑ | ↑ | intact | [39][40][41][42] | [46,47,48,49] | |||||
| whole | ↓ | ↑ | prevention from age-induced cardiomyopathy | [42] | [49] | ||||||||
| endothelium | ↓ | ↑ | ↑ | not available | [11] | [21] | |||||||
| FABP4/5 | whole | ↓ | ↑ | ↑ | ↑ | intact | [13] | [23 | [43] | ,50] | |||
| Meox2 | +/− | :Tcf15 | +/− | endothelium: whole | ↓ | ↑ | ↓ aged | [28] | [38] | ||||
| Rbp-jκ (Notch signal) | endothelium | ⚪ | ↓ | ↑ | ↓ | ↓↓ | [29] | [39] | |||||
| PPARγ | endothelium | →↓ | → | intact (personal observation) | [25] | [35] | |||||||
| VEGF-B | whole | ↓ | ↑ | ↑ | not available | [16] | [26] | ||||||
| FABP3 | whole | ↓ | ↑ | → | ↑ | not available | [44][45] | [51,52] | |||||
| CD36 | cardiomyocyte | → | → | not available | [11] | [21] | |||||||
| cardiomyocyte | ⚪ | ↓ (ex vivo) | ↑ (ex vivo) | intact | [46][47] | [53,54] |
| Ligand | Receptor/Transcription Factor | Target Genes | Target Tissues Influenced by the System | Reference | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PPARγ | CD36 | FABP4 | FABP5 | LPL | GPIHBP1 | ANGPTL4 | LIPG | FATP3 | FATP4 | |||||
| PPARγ | ⚪ | ⚪ | ⚪ | heart, skeletal muscle, adipose tissue | [25][26][27] | [35,36,37] | ||||||||
| Meox2/Tcf15 | ⚪ | ⚪ | ⚪ | ⚪ | ⚪ | ⚪ | heart | [28] | [38] | |||||
| Dll4 | Notch1/N1-ICD/Rbp-jκ | independent | ⚪ | ⚪ | ⚪ | ⚫ | ⚪ | heart, skeletal muscle | [29][30] | [39,40] | ||||
| Apelin | APLNR/phosphorylation of FOXO1 | ⚫ | skeletal muscle | [31] | [41] | |||||||||
| VEGF-B | VEGFR/NPR1 | ⚪ | ⚪ | heart, BAT, skeletal muscle | [16] | [26] | ||||||||
| ANGPTL2 | integrin α5β1 | ⚪ | ⚪ | subcutaneous adipose tissue | [32] | [42] | ||||||||
| 3-HIB | ⚪* | ⚪* | skeletal muscle | [33] | [43] | |||||||||